Microfluidics as a Novel Tool for Biological and Toxicological Assays in Drug Discovery Processes: Focus on Microchip Electrophoresis

 ,
,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
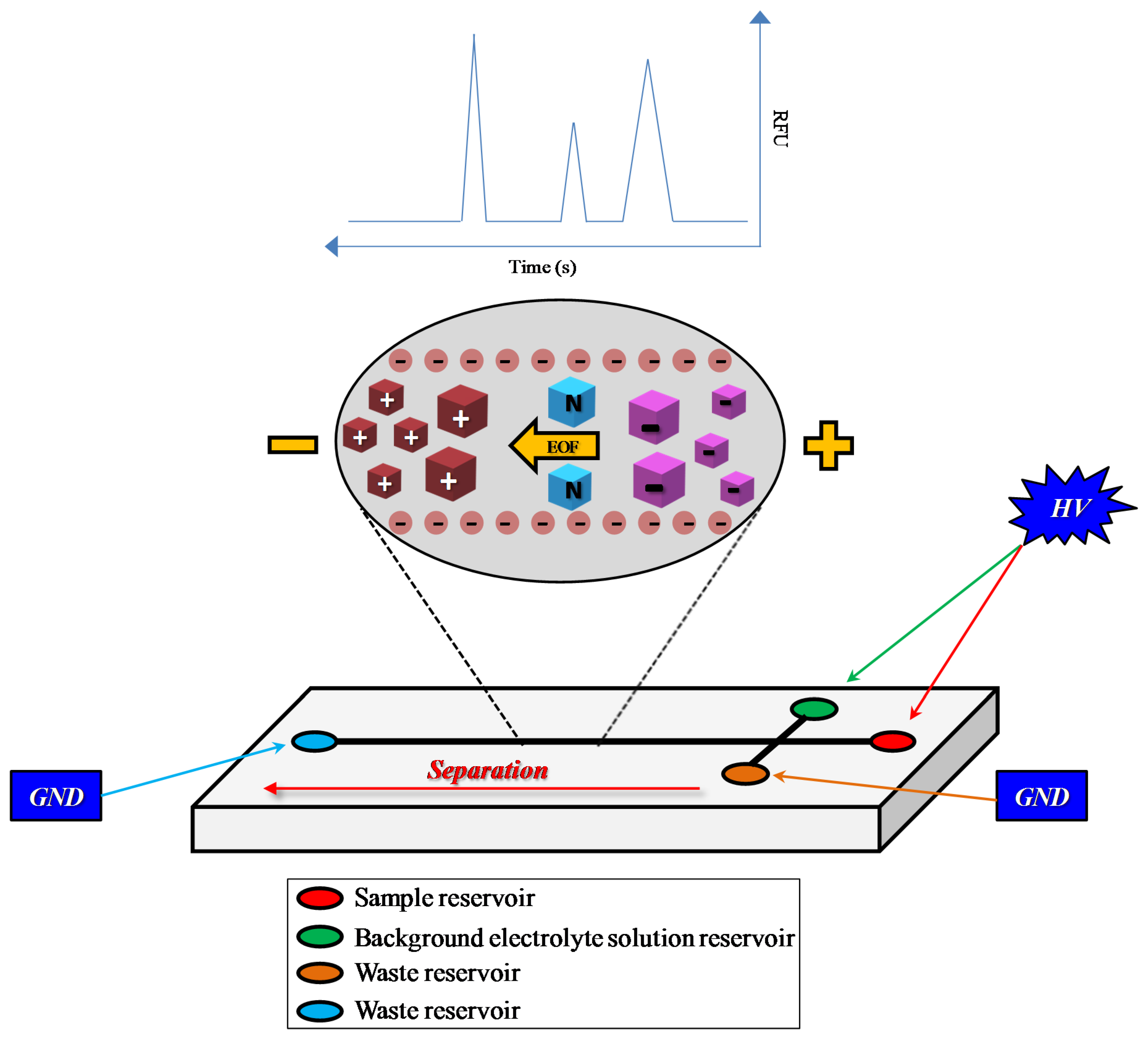
2. Microchip Electrophoresis: Technical Characteristics and Basic Principles
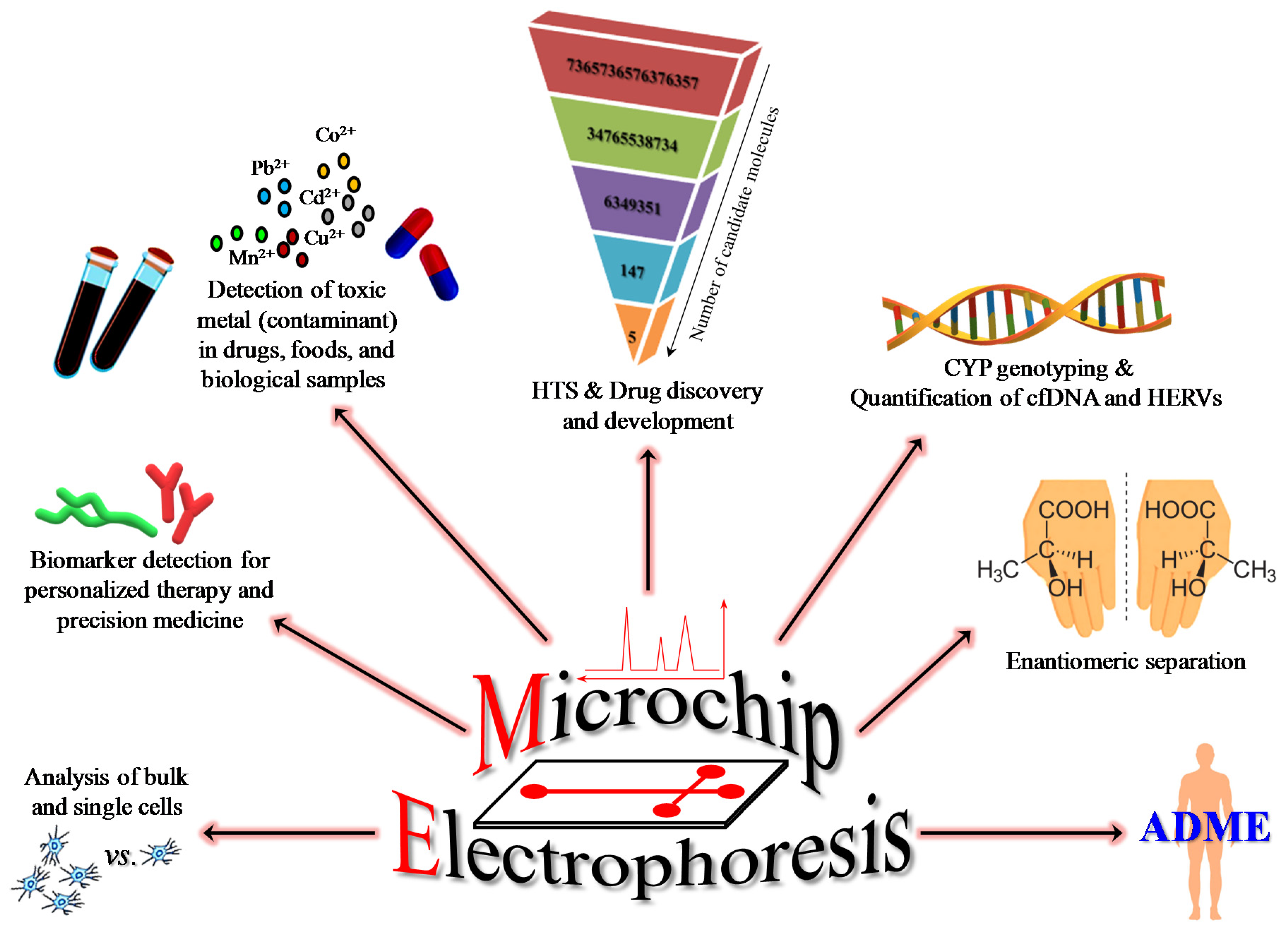
2.1. Role of ME in Drug Discovery and Development and in Preclinical Toxicology
2.2. ME for the Analysis of Bulk and Single Cells
2.3. ME for Biomarker Detection in Personalized Therapy and Precision Medicine
2.4. ME for Separation and Quantification of Neurotransmitters In Vitro and In Vivo
2.5. New Application of ME: The Interesting Case of Carnosine and Its Effects on iNOS Activity and on the Degradation of NO
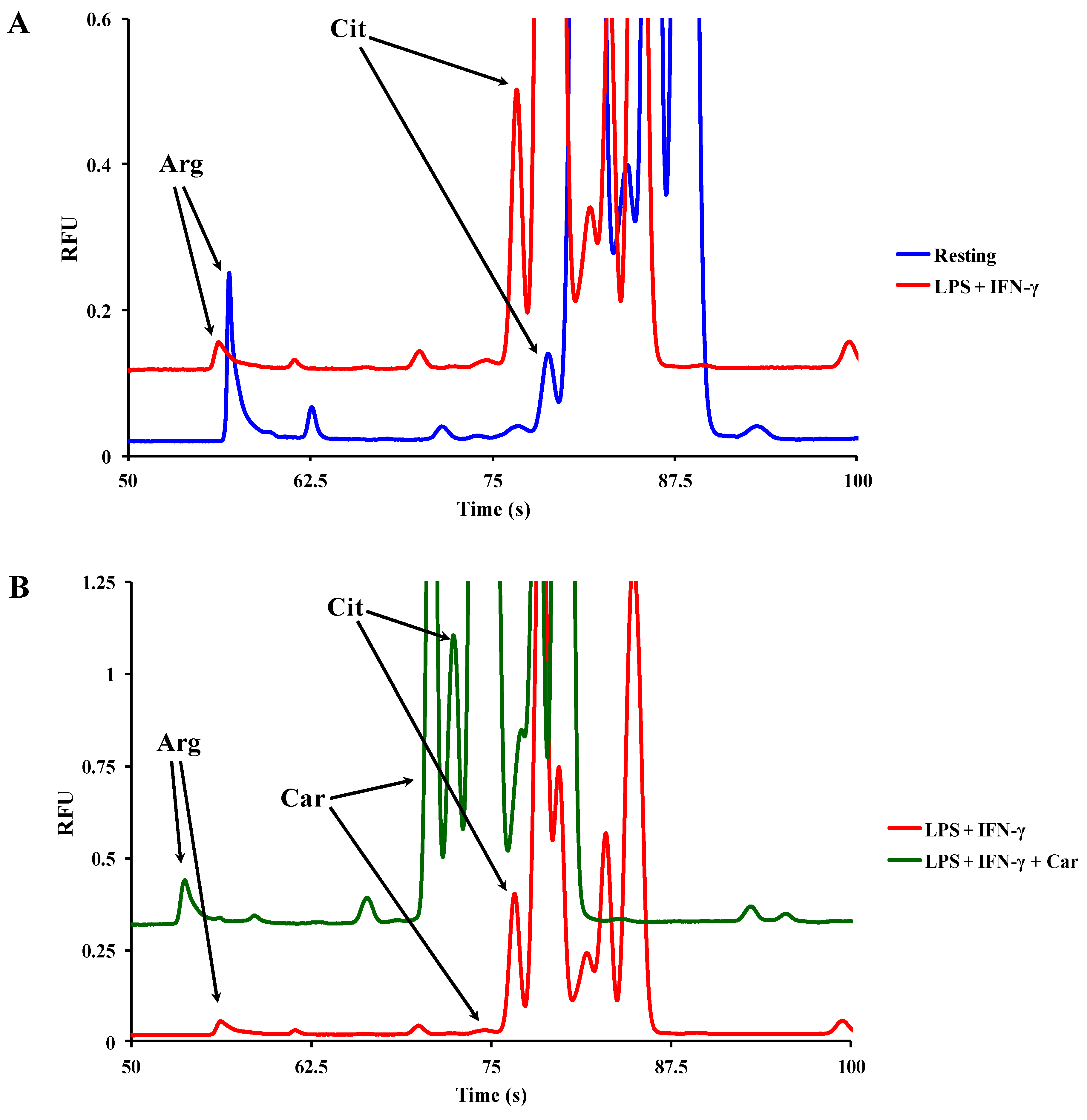
2.5.1. Investigation of the Effects of Carnosine on iNOS Activity in Macrophages by ME-LIF
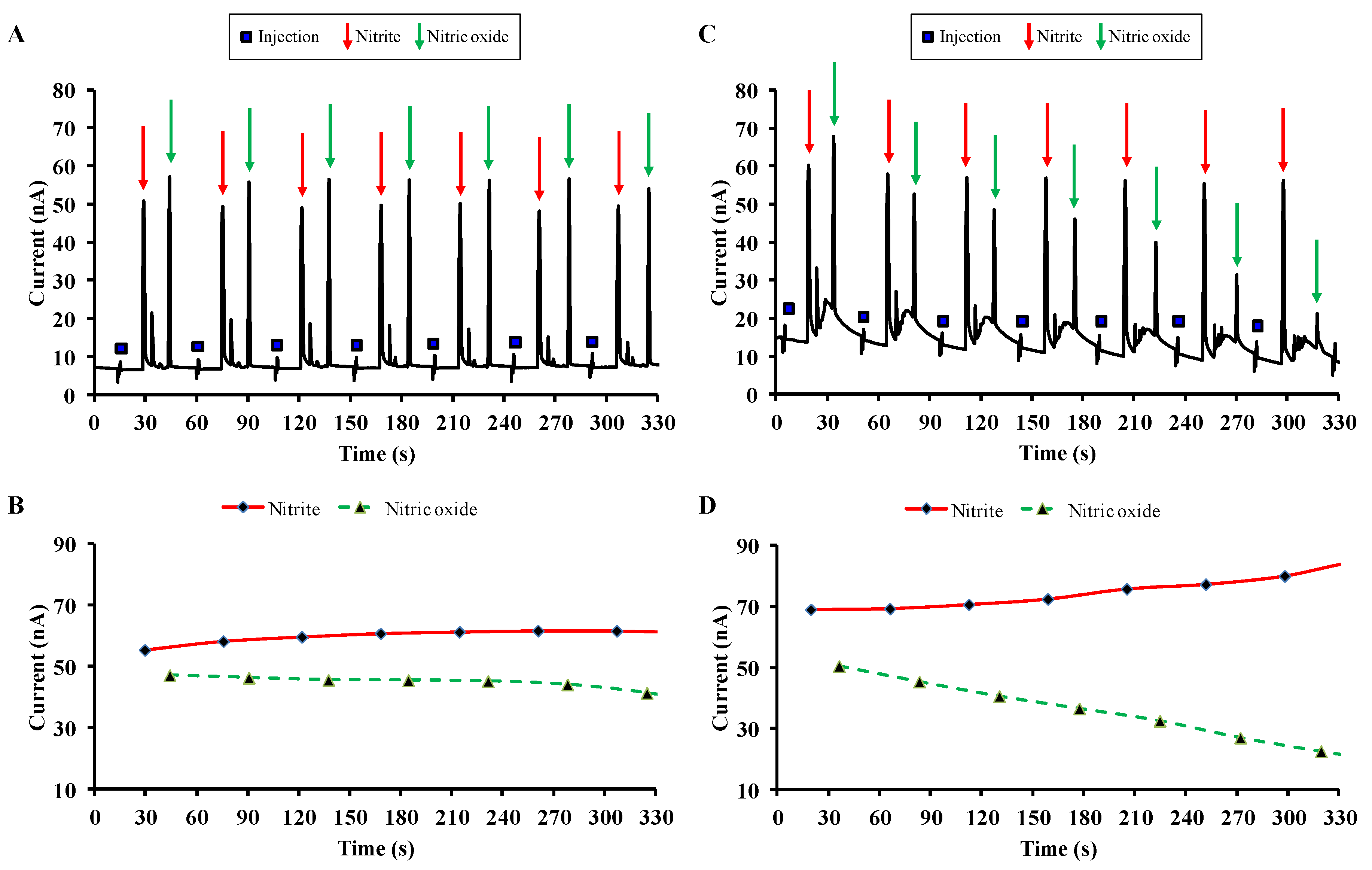
2.5.2. Investigation of Carnosine Effect on NO Degradation by ME-EC
3. Organs-, Organoids-on-a-Chip, and 3D Printing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.C.; Jerman, J.H.; Angell, J.B. A gas chromatographic air analyzer fabricated on a silicon wafer. IEEE Trans. Electron. Devices 1979, 26, 1880–1886. [Google Scholar] [CrossRef]
- Manz, A.; Harrison, D.J.; Verpoorte, E.M.; Fettinger, J.C.; Paulus, A.; Lüdi, H.; Widmer, H.M. Planar chips technology for miniaturization and integration of separation techniques into monitoring systems: Capillary electrophoresis on a chip. J. Chromatogr. A 1992, 593, 253–258. [Google Scholar] [CrossRef]
- Livak-Dahl, E.; Sinn, I.; Burns, M. Microfluidic chemical analysis systems. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 325–353. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by raw 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, R.J.; Wu, M. Microfluidic approaches to fluorescence in situ hybridization (fish) for detecting RNA targets in single cells. In Microfluidic Methods for Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2016; pp. 95–112. [Google Scholar]
- Huang, Y.L.; Segall, J.E.; Wu, M. Microfluidic modeling of the biophysical microenvironment in tumor cell invasion. Lab Chip 2017, 17, 3221–3233. [Google Scholar] [CrossRef]
- Convery, N.; Gadegaard, N. 30 years of microfluidics. Micro Nano Eng. 2019. [Google Scholar] [CrossRef]
- Metto, E.C.; Evans, K.; Barney, P.; Culbertson, A.H.; Gunasekara, D.B.; Caruso, G.; Hulvey, M.K.; da Silva, F.J.A.; Lunte, S.M.; Culbertson, C.T. An integrated microfluidic device for monitoring changes in nitric oxide production in single t-lymphocyte (jurkat) cells. Anal. Chem. 2013, 85, 10188–10195. [Google Scholar] [CrossRef] [Green Version]
- De Campos, R.P.; Siegel, J.M.; Fresta, C.G.; Caruso, G.; da Silva, J.A.; Lunte, S.M. Indirect detection of superoxide in raw 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal. Bioanal. Chem. 2015, 407, 7003–7012. [Google Scholar] [CrossRef]
- Hunter, R.A.; Privett, B.J.; Henley, W.H.; Breed, E.R.; Liang, Z.; Mittal, R.; Yoseph, B.P.; McDunn, J.E.; Burd, E.M.; Coopersmith, C.M.; et al. Microfluidic amperometric sensor for analysis of nitric oxide in whole blood. Anal. Chem. 2013, 85, 6066–6072. [Google Scholar] [CrossRef] [Green Version]
- Vogel, P.A.; Halpin, S.T.; Martin, R.S.; Spence, D.M. Microfluidic transendothelial electrical resistance measurement device that enables blood flow and postgrowth experiments. Anal. Chem. 2011, 83, 4296–4301. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Trouillon, R.; Safina, G.; Ewing, A.G. Chemical analysis of single cells. Anal. Chem. 2011, 83, 4369–4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovarik, M.L.; Gach, P.C.; Ornoff, D.M.; Wang, Y.; Balowski, J.; Farrag, L.; Allbritton, N.L. Micro total analysis systems for cell biology and biochemical assays. Anal. Chem. 2012, 84, 516–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2d vs. 3d cell culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Yu, F.; Hunziker, W.; Choudhury, D. Engineering microfluidic organoid-on-a-chip platforms. Micromachines 2019, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.S.; Sreedharan, S.N.; Patil, S.; Raj, A.T. The potential role of organoids in pathology and oncology research. Pathol. Oncol. Res. 2019, 26, 1353–1354. [Google Scholar] [CrossRef]
- Eribol, P.; Uguz, A.K.; Ulgen, K.O. Screening applications in drug discovery based on microfluidic technology. Biomicrofluidics 2016, 10, 11502. [Google Scholar] [CrossRef] [Green Version]
- Wuethrich, A.; Quirino, J.P. A decade of microchip electrophoresis for clinical diagnostics-a review of 2008–2017. Anal. Chim. Acta 2019, 1045, 42–66. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Fresta, C.G.; Siegel, J.M.; Wijesinghe, M.B.; Lunte, S.M. Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Anal. Bioanal. Chem. 2017, 409, 4529–4538. [Google Scholar] [CrossRef]
- Caruso, G.; Fresta, C.G.; Fidilio, A.; O’Donnell, F.; Musso, N.; Lazzarino, G.; Grasso, M.; Amorini, A.M.; Tascedda, F.; Bucolo, C.; et al. Carnosine decreases pma-induced oxidative stress and inflammation in murine macrophages. Antioxidants 2019, 8, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulvey, M.K.; Frankenfeld, C.N.; Lunte, S.M. Separation and detection of peroxynitrite using microchip electrophoresis with amperometric detection. Anal. Chem. 2010, 82, 1608–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zare, R.N.; Kim, S. Microfluidic platforms for single-cell analysis. Annu. Rev. Biomed. Eng. 2010, 12, 187–201. [Google Scholar] [CrossRef] [Green Version]
- Culbertson, C.T. Single cell analysis on microfluidic devices. Methods Mol. Biol. 2006, 339, 203–216. [Google Scholar] [PubMed]
- Price, A.K.; Culbertson, C.T. Chemical analysis of single mammalian cells with microfluidics. Strategies for culturing, sorting, trapping, and lysing cells and separating their contents on chips. Anal. Chem. 2007, 79, 2614–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovichi, N. Chemical Cytometry: Ultrasensitive Analysis of Single Cells; Lu, C., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Brehm-Stecher, B.F.; Johnson, E.A. Single-cell microbiology: Tools, technologies, and applications. Microbiol. Mol. Biol. Rev. 2004, 68, 538–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Bodovitz, S. Single cell analysis: The new frontier in ‘omics’. Trends Biotechnol. 2010, 28, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Li, S.F.Y. Capillary Electrophoresis: Principles, Practice and Applications; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Ahmadzadeh, H.; Prescott, M.; Muster, N.; Stoyanov, A. Revisiting electroosmotic flow: An important parameter affecting separation in capillary and microchip electrophoresis. Chem. Eng. Commun. 2007, 195, 129–146. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, F.; Zhao, L.; Zhang, J.R.; Zhu, J.J. Measurement of electroosmotic flow in capillary and microchip electrophoresis. J. Chromatogr. A 2007, 1170, 1–8. [Google Scholar] [CrossRef]
- Gomez, F.J.V.; Silva, M.F. Microchip electrophoresis tools for the analysis of small molecules. Methods Mol. Biol. 2019, 1906, 197–206. [Google Scholar]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Vlcková, M. Microchip Electrophoresis Bioanalytical Applications. Ph.D. Thesis, University of Basel, Basel, Switzerland, 2008. [Google Scholar]
- Gomez, F.J.; Silva, M.F. Microchip electrophoresis for wine analysis. Anal. Bioanal. Chem. 2016, 408, 8643–8653. [Google Scholar] [CrossRef]
- Escarpa, A.; Gonzalez, M.C.; Crevillen, A.G.; Blasco, A.J. Ce microchips: An opened gate to food analysis. Electrophoresis 2007, 28, 1002–1011. [Google Scholar] [CrossRef]
- Siegel, J.M.; Schilly, K.M.; Wijesinghe, M.B.; Caruso, G.; Fresta, C.G.; Lunte, S.M. Optimization of a microchip electrophoresis method with electrochemical detection for the determination of nitrite in macrophage cells as an indicator of nitric oxide production. Anal. Methods 2019, 11, 148–156. [Google Scholar] [CrossRef]
- Kristoff, C.J.; Bwanali, L.; Veltri, L.M.; Gautam, G.P.; Rutto, P.K.; Newton, E.O.; Holland, L.A. Challenging bioanalyses with capillary electrophoresis. Anal. Chem. 2020, 92, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Creamer, J.S.; Oborny, N.J.; Lunte, S.M. Recent advances in the analysis of therapeutic proteins by capillary and microchip electrophoresis. Anal. Methods 2014, 6, 5427–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.X.; Manz, A. Narrow sample channel injectors for capillary electrophoresis on microchips. Anal. Chem. 2001, 73, 2656–2662. [Google Scholar] [CrossRef]
- Jacobson, S.C.; Hergenroder, R.; Koutny, L.B.; Warmack, R.; Ramsey, J.M. Effects of injection schemes and column geometry on the performance of microchip electrophoresis devices. Anal. Chem. 1994, 66, 1107–1113. [Google Scholar] [CrossRef]
- Jacobson, S.C.; Koutny, L.B.; Hergenroeder, R.; Moore, A.W.; Ramsey, J.M. Microchip capillary electrophoresis with an integrated postcolumn reactor. Anal. Chem. 1994, 66, 3472–3476. [Google Scholar] [CrossRef]
- Lee, N.Y.; Yamada, M.; Seki, M. Pressure-driven sample injection with quantitative liquid dispensing for on-chip electrophoresis. Anal. Sci. 2004, 20, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Nuchtavorn, N.; Suntornsuk, W.; Lunte, S.M.; Suntornsuk, L. Recent applications of microchip electrophoresis to biomedical analysis. J. Pharm. Biomed. Anal. 2015, 113, 72–96. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.; Malavaki, C.; Tzanakakis, G.; Tsatsakis, A. Principles and applications of capillary electrophoresis and microchips in forensic and clinical toxicology. Toxicol. Lett. 2008, S1. [Google Scholar] [CrossRef]
- Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortino, M.A.; et al. Carnosine prevents aβ-induced oxidative stress and inflammation in microglial cells: A key role of tgf-β1. Cells 2019, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilly, K.M.; Gunawardhana, S.M.; Wijesinghe, M.B.; Lunte, S.M. Biological applications of microchip electrophoresis with amperometric detection: In vivo monitoring and cell analysis. Anal. Bioanal. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Breadmore, M.C. Capillary and microchip electrophoresis: Challenging the common conceptions. J. Chromatogr. A 2012, 1221, 42–55. [Google Scholar] [CrossRef]
- Ouimet, C.M.; D’Amico, C.I.; Kennedy, R.T. Advances in capillary electrophoresis and the implications for drug discovery. Expert. Opin. Drug Discov. 2017, 12, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Perrin, D.; Fremaux, C.; Shutes, A. Capillary microfluidic electrophoretic mobility shift assays: Application to enzymatic assays in drug discovery. Expert. Opin. Drug Discov. 2010, 5, 51–63. [Google Scholar] [CrossRef]
- Pan, J.; Chen, Z.; Yao, M.; Li, X.; Li, Y.; Sun, D.; Yu, Y. A two-electrode system-based electrochemiluminescence detection for microfluidic capillary electrophoresis and its application in pharmaceutical analysis. Luminescence 2014, 29, 427–432. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Z.; Yu, Y.; Yang, J.; Pan, J. Determination of sulfonamides in pharmaceuticals and rabbit plasma by microchip electrophoresis with led-if detection. Chromatographia 2013, 76, 821–829. [Google Scholar] [CrossRef]
- Nordman, N.; Sikanen, T.; Moilanen, M.E.; Aura, S.; Kotiaho, T.; Franssila, S.; Kostiainen, R. Rapid and sensitive drug metabolism studies by su-8 microchip capillary electrophoresis-electrospray ionization mass spectrometry. J. Chromatogr. A 2011, 1218, 739–745. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: It’s the same thing. Only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, X.C.; Zhang, W.; Pan, J.B.; Chen, Z.G. A facile light-emitting-diode induced fluorescence detector coupled to an integrated microfluidic device for microchip electrophoresis. Talanta 2011, 84, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Toxicity of metal compounds: Knowledge and myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef] [Green Version]
- Rafique, S.; Idrees, M.; Nasim, A.; Akbar, H.; Athar, A. Transition metal complexes as potential therapeutic agents. Biotechnol. Mol. Biol. Rev. 2010, 5, 38–45. [Google Scholar]
- Jacobson, S.C.; Moore, A.W.; Ramsey, J.M. Fused quartz substrates for microchip electrophoresis. Anal. Chem. 1995, 67, 2059–2063. [Google Scholar] [CrossRef]
- Faure, K.; Loughran, M.; Glennon, J.D. Metal Complex Speciation on-Chip. In Proceedings of the Microfluidics, BioMEMS, and Medical Microsystems IV, San Jose, CA, USA, 21–27 January 2006; International Society for Optics and Photonics: Bellingham, DC, USA, 2006; p. 61120. [Google Scholar]
- Faure, K.; Pravda, M.; Glennon, J.D. Microchip electrophoresis: A new platform for metal speciation. Anal. Lett. 2006, 39, 435–449. [Google Scholar] [CrossRef]
- Rose, M.; Fernandes, A. Persistent organic pollutants and toxic metals in foods. Qual. Assur. Saf. Crop. Foods 2014, 6, 115–116. [Google Scholar]
- Liu, B.; Zhang, Y.; Mayer, D.; Krause, H.J.; Jin, Q.; Zhao, J.; Offenhausser, A.; Xu, Y. Determination of heavy metal ions by microchip capillary electrophoresis coupled with contactless conductivity detection. Electrophoresis 2012, 33, 1247–1250. [Google Scholar] [CrossRef]
- Deng, G.; Collins, G.E. Nonaqueous based microchip separation of toxic metal ions using 2-(5-bromo-2-pyridylazo)-5-(n-propyl-n-sulfopropylamino) phenol. J. Chromatogr. A 2003, 989, 311–316. [Google Scholar] [CrossRef]
- Prasanna, R.; Jayasri, V.; Yasmeen, R.; Chowdary, H.; Satyanandam, S. Significance of pharmaceutical excipients-a review. J. Innov. Trends Pharm. Sci. 2011, 2, 191–201. [Google Scholar]
- Troška, P.; Poboży, E.; Némethová, Z.; Masár, M. Determination of commonly used excipients in pharmaceutical preparations by microchip electrophoresis with conductivity detection. Chromatographia 2019, 82, 741–748. [Google Scholar] [CrossRef]
- Perrin, D.; Fremaux, C.; Scheer, A. Assay development and screening of a serine/threonine kinase in an on-chip mode using caliper nanofluidics technology. J. Biomol. Screen. 2006, 11, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Perrin, D.; Martin, T.; Cambet, Y.; Fremaux, C.; Scheer, A. Overcoming the hurdle of fluorescent compounds in kinase screening: A case study. Assay Drug Dev. Technol. 2006, 4, 185–196. [Google Scholar] [CrossRef]
- Pommereau, A.; Pap, E.; Kannt, A. Two simple and generic antibody-independent kinase assays: Comparison of a bioluminescent and a microfluidic assay format. J. Biomol. Screen. 2004, 9, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.; Reardon, H.; Trinh, V.; Li, E.; Farinas, J. Comparison of on-chip and off-chip microfluidic kinase assay formats. Assay Drug Dev. Technol. 2004, 2, 121–129. [Google Scholar] [CrossRef]
- Liu, Y.; Gerber, R.; Wu, J.; Tsuruda, T.; McCarter, J.D. High-throughput assays for sirtuin enzymes: A microfluidic mobility shift assay and a bioluminescence assay. Anal. Biochem. 2008, 378, 53–59. [Google Scholar] [CrossRef]
- Perrin, D.; Fremaux, C.; Besson, D.; Sauer, W.H.; Scheer, A. A microfluidics-based mobility shift assay to discover new tyrosine phosphatase inhibitors. J. Biomol. Screen. 2006, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourtounis, J.; Falgueyret, J.P.; Sayegh, C.E. Assessing protein-RNA interactions using microfluidic capillary mobility shift assays. Anal. Biochem. 2011, 411, 161–163. [Google Scholar] [CrossRef]
- Ludwig, M.; Kohler, F.; Belder, D. High-speed chiral separations on a microchip with UV detection. Electrophoresis 2003, 24, 3233–3238. [Google Scholar] [CrossRef] [PubMed]
- Correia, I.R. Stability of igg isotypes in serum. MAbs 2010, 2, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Piparia, R.; Ouellette, D.; Stine, W.B.; Grinnell, C.; Tarcsa, E.; Radziejewski, C.; Correia, I. A high throughput capillary electrophoresis method to obtain pharmacokinetics and quality attributes of a therapeutic molecule in circulation. MAbs 2012, 4, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Huang, W.; Cheng, J. Determination of neurotransmitters in pc 12 cells by microchip electrophoresis with fluorescence detection. Electrophoresis 2007, 28, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Huang, W.; Cheng, J. Analysis of amino acids in human vascular endothelial (ecv-304) cells by microchip electrophoresis with fluorescence detection. J. Sep. Sci. 2008, 31, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- ScottáMartin, R. In celebration of the 60th birthday of 2 microfluidics pioneers: Professor Susan Lunte and professor James Landers. Anal. Methods 2018, 10, 3433–3435. [Google Scholar]
- Caruso, G.; Spampinato, S.F.; Cardaci, V.; Caraci, F.; Sortino, M.A.; Merlo, S. Beta-amyloid and oxidative stress: Perspectives in drug development. Curr. Pharm. Des. 2019, 25, 4771–4781. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Mainz, E.R.; Gunasekara, D.B.; Caruso, G.; Jensen, D.T.; Hulvey, M.K.; da Silva, J.A.F.; Metto, E.C.; Culbertson, A.H.; Culbertson, C.T.; Lunte, S.M. Monitoring intracellular nitric oxide production using microchip electrophoresis and laser-induced fluorescence detection. Anal. Methods 2012, 4, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Lunte, S.M.; Gunasekara, D.B.; Metto, E.C.; Hulvey, M.K.; Mainz, E.R.; Caruso, G.; Da Silva, J.A.F.; Jensen, D.T.; Culbertson, A.H.; Grigsby, R.J. Microchip Electrophoresis Devices for the Detection of Nitric Oxide: Comparison of Bulk Cell and Single Cell Analysis. In Proceedings of the 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Seattle, WA, USA, 2–6 October 2011; MicroTAS: Palm Springs, CA, USA, 2011; pp. 1728–1730. [Google Scholar]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gunasekara, D.B.; Siegel, J.M.; Caruso, G.; Hulvey, M.K.; Lunte, S.M. Microchip electrophoresis with amperometric detection method for profiling cellular nitrosative stress markers. Analyst 2014, 139, 3265–3273. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Fresta, C.G.; Martinez-Becerra, F.; Antonio, L.; Johnson, R.T.; de Campos, R.P.S.; Siegel, J.M.; Wijesinghe, M.B.; Lazzarino, G.; Lunte, S.M. Carnosine modulates nitric oxide in stimulated murine raw 264.7 macrophages. Mol. Cell. Biochem. 2017, 431, 197–210. [Google Scholar] [CrossRef]
- Schmid, A.; Kortmann, H.; Dittrich, P.S.; Blank, L.M. Chemical and biological single cell analysis. Curr. Opin. Biotechnol. 2010, 21, 12–20. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.A.; Culbertson, C.T.; Jacobson, S.C.; Allbritton, N.L.; Sims, C.E.; Ramsey, J.M. Microfluidic devices for the high-throughput chemical analysis of cells. Anal. Chem. 2003, 75, 5646–5655. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Huang, Y.; Zhao, J.; Li, S.; Liu, R.; Zhao, S. Quantification of glutathione in single cells from rat liver by microchip electrophoresis with chemiluminescence detection. Talanta 2018, 179, 466–471. [Google Scholar] [CrossRef]
- Ling, Y.Y.; Yin, X.F.; Fang, Z.L. Simultaneous determination of glutathione and reactive oxygen species in individual cells by microchip electrophoresis. Electrophoresis 2005, 26, 4759–4766. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, S.; Hu, H.; Liu, Y.M. A microchip electrophoresis-mass spectrometric platform with double cell lysis nano-electrodes for automated single cell analysis. J. Chromatogr. A 2016, 1451, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, Q.; Chen, P.; Li, Z.; Chen, Z.; Tang, B. Consecutive gated injection-based microchip electrophoresis for simultaneous quantitation of superoxide anion and nitric oxide in single pc-12 cells. Anal. Chem. 2016, 88, 930–936. [Google Scholar] [CrossRef]
- Mizukami, K.; Chang, H.S.; Yabuki, A.; Kawamichi, T.; Hossain, M.A.; Rahman, M.M.; Uddin, M.M.; Yamato, O. Rapid genotyping assays for the 4-base pair deletion of canine mdr1/abcb1 gene and low frequency of the mutant allele in border collie dogs. J. Vet. Diagn. Investig. 2012, 24, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Klintzsch, S.; Meerkamp, K.; Doring, B.; Geyer, J. Detection of the nt230[del4] mdr1 mutation in dogs by a fluorogenic 5′ nuclease taqman allelic discrimination method. Vet. J. 2010, 185, 272–277. [Google Scholar] [CrossRef]
- Poe, B.L.; Haverstick, D.M.; Landers, J.P. Warfarin genotyping in a single pcr reaction for microchip electrophoresis. Clin. Chem. 2012, 58, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, J.; Takinami, Y.; Ueki, M.; Kimura-Kataoka, K.; Yasuda, T.; Takeshita, H. Circulating cell-free DNA fragment analysis by microchip electrophoresis and its relationship with dnase i in cardiac diseases. Clin. Chim. Acta 2019, 497, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Lang, A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin. Rev. Allergy Immunol. 2010, 39, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human endogenous retroviruses in neurological diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Cakmak Guner, B.; Karlik, E.; Marakli, S.; Gozukirmizi, N. Detection of herv-k6 and herv-k11 transpositions in the human genome. Biomed. Rep. 2018, 9, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, B.; Boller, K.; Reuter, A.; Schnierle, B.S. Characterization of the human endogenous retrovirus k gag protein: Identification of protease cleavage sites. Retrovirology 2011, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Lin, X.; Lin, L.; Yi, L.; Li, H.; Lin, J.M. A comparative study of three different nucleic acid amplification techniques combined with microchip electrophoresis for hpv16 e6/e7 mrna detection. Analyst 2015, 140, 6736–6741. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ni, K.; Zhou, G. Microchip electrophoresis coupled with multiplex polymerase chain reaction for typing multiple single nucleotide polymorphisms simultaneously. Chin. J. Anal. Chem. 2006, 34, 1389–1394. [Google Scholar] [CrossRef]
- Kobylecki, C.J.; Jakobsen, K.D.; Hansen, T.; Jakobsen, I.V.; Rasmussen, H.B.; Werge, T. Cyp2d6 genotype predicts antipsychotic side effects in schizophrenia inpatients: A retrospective matched case-control study. Neuropsychobiology 2009, 59, 222–226. [Google Scholar] [CrossRef]
- van Gelder, T.; Hesselink, D.A. Dosing tacrolimus based on cyp3a5 genotype: Will it improve clinical outcome? Clin Pharm. 2010, 87, 640–641. [Google Scholar] [CrossRef]
- Qu, L.; Lu, Y.; Ying, M.; Li, B.; Weng, C.; Xie, Z.; Liang, L.; Lin, C.; Yang, X.; Feng, S.; et al. Tacrolimus dose requirement based on the cyp3a5 genotype in renal transplant patients. Oncotarget 2017, 8, 81285–81294. [Google Scholar] [CrossRef] [Green Version]
- Calvo, P.L.; Serpe, L.; Brunati, A.; Nonnato, A.; Bongioanni, D.; Olio, D.D.; Pinon, M.; Ferretti, C.; Tandoi, F.; Carbonaro, G.; et al. Donor cyp3a5 genotype influences tacrolimus disposition on the first day after paediatric liver transplantation. Br. J. Clin. Pharm. 2017, 83, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, C.; Li, J.; Wang, X.; Zhu, G.; Chen, X.; Bi, H.; Huang, M. Effect of genetic polymorphisms of cyp3a5 and mdr1 on cyclosporine concentration during the early stage after renal transplantation in Chinese patients co-treated with diltiazem. Eur. J. Clin. Pharm. 2009, 65, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Anglicheau, D.; Thervet, E.; Etienne, I.; Hurault De Ligny, B.; Le Meur, Y.; Touchard, G.; Buchler, M.; Laurent-Puig, P.; Tregouet, D.; Beaune, P.; et al. Cyp3a5 and mdr1 genetic polymorphisms and cyclosporine pharmacokinetics after renal transplantation. Clin Pharm. 2004, 75, 422–433. [Google Scholar] [CrossRef]
- Minarik, M.; Kopeckova, M.; Gassman, M.; Osmancik, P.; Benesova, L. Rapid testing of clopidogrel resistance by genotyping of cyp2c19 and cyp2c9 polymorphisms using denaturing on-chip capillary electrophoresis. Electrophoresis 2012, 33, 1306–1310. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.I.; Desmulliez, M.P. Lab-on-a-chip based immunosensor principles and technologies for the detection of cardiac biomarkers: A review. Lab Chip 2011, 11, 569–595. [Google Scholar] [CrossRef]
- Ruecha, N.; Siangproh, W.; Chailapakul, O. A fast and highly sensitive detection of cholesterol using polymer microfluidic devices and amperometric system. Talanta 2011, 84, 1323–1328. [Google Scholar] [CrossRef]
- Wang, H.; Wang, D.; Wang, J.; Wang, H.; Gu, J.; Han, C.; Jin, Q.; Xu, B.; He, C.; Cao, L.; et al. Application of poly(dimethylsiloxane)/glass microchip for fast electrophoretic separation of serum small, dense low-density lipoprotein. J. Chromatogr. A 2009, 1216, 6343–6347. [Google Scholar] [CrossRef]
- Gibson, L.R.; Bohn, P.W. Non-aqueous microchip electrophoresis for characterization of lipid biomarkers. Interface Focus 2013, 3, 20120096. [Google Scholar] [CrossRef]
- Caruso, G.; Fresta, C.G.; Lazzarino, G.; Distefano, D.A.; Parlascino, P.; Lunte, S.M.; Lazzarino, G.; Caraci, F. Sub-toxic human amylin fragment concentrations promote the survival and proliferation of sh-sy5y cells via the release of vegf and hspb5 from endothelial rbe4 cells. Int. J. Mol. Sci. 2018, 19, 3659. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Distefano, D.A.; Parlascino, P.; Fresta, C.G.; Lazzarino, G.; Lunte, S.M.; Nicoletti, V.G. Receptor-mediated toxicity of human amylin fragment aggregated by short-and long-term incubations with copper ions. Mol. Cell Biochem. 2017, 425, 85–93. [Google Scholar] [CrossRef]
- Porsch-Ozcurumez, M.; Kischel, N.; Priebe, H.; Splettstosser, W.; Finke, E.J.; Grunow, R. Comparison of enzyme-linked immunosorbent assay, western blotting, microagglutination, indirect immunofluorescence assay, and flow cytometry for serological diagnosis of tularemia. Clin. Diagn. Lab. Immunol. 2004, 11, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamadi, M.R.; Svobodova, Z.; Verpillot, R.; Esselmann, H.; Wiltfang, J.; Otto, M.; Taverna, M.; Bilkova, Z.; Viovy, J.L. Microchip electrophoresis profiling of abeta peptides in the cerebrospinal fluid of patients with Alzheimer’s disease. Anal. Chem. 2010, 82, 7611–7617. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mallory, T.; Satomura, S. Afp-l3: A new generation of tumor marker for hepatocellular carcinoma. Clin. Chim. Acta 2001, 313, 15–19. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hosaka, T.; Ikeda, K.; Seko, Y.; Kawamura, Y.; Sezaki, H.; Akuta, N.; Suzuki, F.; Suzuki, Y.; Saitoh, S.; et al. Highly sensitive afp-l3% assay is useful for predicting recurrence of hepatocellular carcinoma after curative treatment pre-and postoperatively. Hepatol. Res. 2011, 41, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; Wellner, E. Detection of cerebral spinal fluid-associated chemokines in birth traumatized premature babies by chip-based immunoaffinity CE. Electrophoresis 2013, 34, 1530–1538. [Google Scholar] [CrossRef] [Green Version]
- Kailemia, M.J.; Ruhaak, L.R.; Lebrilla, C.B.; Amster, I.J. Oligosaccharide analysis by mass spectrometry: A review of recent developments. Anal. Chem. 2014, 86, 196–212. [Google Scholar] [CrossRef] [Green Version]
- Mitra, I.; Alley, W.R., Jr.; Goetz, J.A.; Vasseur, J.A.; Novotny, M.V.; Jacobson, S.C. Comparative profiling of n-glycans isolated from serum samples of ovarian cancer patients and analyzed by microchip electrophoresis. J. Proteome. Res. 2013, 12, 4490–4496. [Google Scholar] [CrossRef] [Green Version]
- Duarte, G.F., Jr.; Lobo, E.O., Jr.; Medeiros, I., Jr.; da Silva, J.A.F.; do Lago, C.L.; Coltro, W.K.T. Separation of carbohydrates on electrophoresis microchips with controlled electrolysis. Electrophoresis 2019, 40, 693–698. [Google Scholar] [CrossRef]
- Maeda, E.; Kataoka, M.; Hino, M.; Kajimoto, K.; Kaji, N.; Tokeshi, M.; Kido, J.; Shinohara, Y.; Baba, Y. Determination of human blood glucose levels using microchip electrophoresis. Electrophoresis 2007, 28, 2927–2933. [Google Scholar] [CrossRef]
- Saylor, R.A.; Lunte, S.M. A review of microdialysis coupled to microchip electrophoresis for monitoring biological events. J. Chromatogr. A 2015, 1382, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, C.F.; Puglia, M.P.; Zucca, S. Focus on: Neurotransmitter systems. Alcohol Res. Health 2011, 34, 106–120. [Google Scholar] [PubMed]
- Li, X.; Hu, H.; Zhao, S.; Liu, Y.M. Microfluidic platform with in-chip electrophoresis coupled to mass spectrometry for monitoring neurochemical release from nerve cells. Anal. Chem. 2016, 88, 5338–5344. [Google Scholar] [CrossRef] [Green Version]
- Vlckova, M.; Schwarz, M.A. Determination of cationic neurotransmitters and metabolites in brain homogenates by microchip electrophoresis and carbon nanotube-modified amperometry. J. Chromatogr. A 2007, 1142, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Wang, G.; Chen, W.; Li, Y.; Zhang, Y.; He, P.; Wang, Q. Sensitive determination of neurotransmitters in urine by microchip electrophoresis with multiple-concentration approaches combining field-amplified and reversed-field stacking. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1025, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Roman, G.T.; Perry, M.L.; Kennedy, R.T. Microfluidic chip for high efficiency electrophoretic analysis of segmented flow from a microdialysis probe and in vivo chemical monitoring. Anal. Chem. 2009, 81, 9072–9078. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Steven, S.; Daiber, A. Organic nitrates: Update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vasc. Pharm. 2014, 63, 105–113. [Google Scholar] [CrossRef]
- Scott, D.E.; Willis, S.D.; Gabbert, S.; Johnson, D.; Naylor, E.; Janle, E.M.; Krichevsky, J.E.; Lunte, C.E.; Lunte, S.M. Development of an on-animal separation-based sensor for monitoring drug metabolism in freely roaming sheep. Analyst 2015, 140, 3820–3829. [Google Scholar] [CrossRef] [Green Version]
- Sandlin, Z.D.; Shou, M.; Shackman, J.G.; Kennedy, R.T. Microfluidic electrophoresis chip coupled to microdialysis for in vivo monitoring of amino acid neurotransmitters. Anal. Chem. 2005, 77, 7702–7708. [Google Scholar] [CrossRef]
- Gunawardhana, S.M.; Bulgakova, G.A.; Barybin, A.M.; Thomas, S.R.; Lunte, S.M. Progress toward the development of a microchip electrophoresis separation-based sensor with electrochemical detection for on-line in vivo monitoring of catecholamines. Analyst 2020, 145, 1768–1776. [Google Scholar] [CrossRef]
- Sibbitts, J.; Sellens, K.A.; Jia, S.; Klasner, S.A.; Culbertson, C.T. Cellular analysis using microfluidics. Anal. Chem. 2018, 90, 65–85. [Google Scholar] [CrossRef]
- Bitar, M.S.; Wahid, S.; Mustafa, S.; Al-Saleh, E.; Dhaunsi, G.S.; Al-Mulla, F. Nitric oxide dynamics and endothelial dysfunction in type ii model of genetic diabetes. Eur. J. Pharm. 2005, 511, 53–64. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresta, C.G.; Fidilio, A.; Lazzarino, G.; Musso, N.; Grasso, M.; Merlo, S.; Amorini, A.M.; Bucolo, C.; Tavazzi, B.; Lazzarino, G.; et al. Modulation of pro-oxidant and pro-inflammatory activities of m1 macrophages by the natural dipeptide carnosine. Int. J. Mol. Sci. 2020, 21, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linz, T.H.; Snyder, C.M.; Lunte, S.M. Optimization of the separation of nda-derivatized methylarginines by capillary and microchip electrophoresis. J. Lab. Autom. 2012, 17, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G. Basi Molecolari dei DCP (Disoridni Conformazionali Proteici) a Carico del Sistema Nervoso: Condizioni Microambientali e Interrelazioni Cellulari. Ph.D. Thesis, University of Catania, Catania, Italy, 2014. [Google Scholar]
- Watson, D.E.; Hunziker, R.; Wikswo, J.P. Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp. Biol. Med. 2017, 242, 1559–1572. [Google Scholar] [CrossRef] [Green Version]
- Augustyniak, J.; Bertero, A.; Coccini, T.; Baderna, D.; Buzanska, L.; Caloni, F. Organoids are promising tools for species-specific in vitro toxicological studies. J. Appl. Toxicol. 2019, 39, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Sin, A.; Chin, K.C.; Jamil, M.F.; Kostov, Y.; Rao, G.; Shuler, M.L. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol. Prog. 2004, 20, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Viravaidya, K.; Sin, A.; Shuler, M.L. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol. Prog. 2004, 20, 316–323. [Google Scholar] [CrossRef]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharm. 2018, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, C.; Jiang, L.; Qin, J. A 3d human lung-on-a-chip model for nanotoxicity testing. Toxicol. Res. 2018, 7, 1048–1060. [Google Scholar] [CrossRef] [Green Version]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [Green Version]
- Bein, A.; Shin, W.; Jalili-Firoozinezhad, S.; Park, M.H.; Sontheimer-Phelps, A.; Tovaglieri, A.; Chalkiadaki, A.; Kim, H.J.; Ingber, D.E. Microfluidic organ-on-a-chip models of human intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Van der Helm, M.W.; Henry, O.Y.F.; Bein, A.; Hamkins-Indik, T.; Cronce, M.J.; Leineweber, W.D.; Odijk, M.; van der Meer, A.D.; Eijkel, J.C.T.; Ingber, D.E.; et al. Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy. Lab Chip 2019, 19, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ingber, D.E. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gunasekara, D.B.; Reed, M.I.; DiSalvo, M.; Bultman, S.J.; Sims, C.E.; Magness, S.T.; Allbritton, N.L. A microengineered collagen scaffold for generating a polarized crypt-villus architecture of human small intestinal epithelium. Biomaterials 2017, 128, 44–55. [Google Scholar] [CrossRef]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (mubbb). Lab Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Neely, M.D.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M.; et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 54124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. Bbb on chip: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Byun, W.Y.; Frank, A.; Massaro-Giordano, M.; Lee, V.; Bunya, V.Y.; Huh, D. Human blinking ‘eye-on-a-chip’. Investig. Ophthalmol. Vis. Sci 2016, 57, 3872. [Google Scholar]
- Abdalkader, R.; Kamei, K.I. Multi-corneal barrier-on-a-chip to recapitulate eye blinking shear stress forces. Lab Chip 2020. [Google Scholar] [CrossRef] [PubMed]
- Abaci, H.E.; Gledhill, K.; Guo, Z.; Christiano, A.M.; Shuler, M.L. Pumpless microfluidic platform for drug testing on human skin equivalents. Lab Chip 2015, 15, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gledhill, K.; Guo, Z.; Umegaki-Arao, N.; Higgins, C.A.; Itoh, M.; Christiano, A.M. Melanin transfer in human 3d skin equivalents generated exclusively from induced pluripotent stem cells. PLoS ONE 2015, 10, e0136713. [Google Scholar] [CrossRef]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef]
- Powers, M.J.; Domansky, K.; Kaazempur-Mofrad, M.R.; Kalezi, A.; Capitano, A.; Upadhyaya, A.; Kurzawski, P.; Wack, K.E.; Stolz, D.B.; Kamm, R.; et al. A microfabricated array bioreactor for perfused 3d liver culture. Biotechnol. Bioeng. 2002, 78, 257–269. [Google Scholar] [CrossRef]
- Nakao, Y.; Kimura, H.; Sakai, Y.; Fujii, T. Bile canaliculi formation by aligning rat primary hepatocytes in a microfluidic device. Biomicrofluidics 2011, 5, 22212. [Google Scholar] [CrossRef] [Green Version]
- Van Midwoud, P.M.; Verpoorte, E.; Groothuis, G.M. Microfluidic devices for in vitro studies on liver drug metabolism and toxicity. Integr. Biol. 2011, 3, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Suh, K.Y. A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab Chip 2010, 10, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Wang, Y.; Chen, W.; Li, Z.; Su, W.; Guo, Y.; Deng, P.; Qin, J. Engineering human islet organoids from ipscs using an organ-on-chip platform. Lab Chip 2019, 19, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.K.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat. Commun. 2019, 10, 3124. [Google Scholar]
- Loskill, P.; Sezhian, T.; Tharp, K.M.; Lee-Montiel, F.T.; Jeeawoody, S.; Reese, W.M.; Zushin, P.H.; Stahl, A.; Healy, K.E. Wat-on-a-chip: A physiologically relevant microfluidic system incorporating white adipose tissue. Lab Chip 2017, 17, 1645–1654. [Google Scholar] [CrossRef]
- Liu, Y.; Kongsuphol, P.; Chiam, S.Y.; Zhang, Q.X.; Gourikutty, S.B.N.; Saha, S.; Biswas, S.K.; Ramadan, Q. Adipose-on-a-chip: A dynamic microphysiological in vitro model of the human adipose for immune-metabolic analysis in type II diabetes. Lab Chip 2019, 19, 241–253. [Google Scholar] [CrossRef]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [Green Version]
- Mannhardt, I.; Breckwoldt, K.; Letuffe-Breniere, D.; Schaaf, S.; Schulz, H.; Neuber, C.; Benzin, A.; Werner, T.; Eder, A.; Schulze, T.; et al. Human engineered heart tissue: Analysis of contractile force. Stem Cell Rep. 2016, 7, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3d cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef]
- De Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human brain organoids on a chip reveal the physics of folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef]
- Qu, Y.; Han, B.; Gao, B.; Bose, S.; Gong, Y.; Wawrowsky, K.; Giuliano, A.E.; Sareen, D.; Cui, X. Differentiation of human induced pluripotent stem cells to mammary-like organoids. Stem Cell Rep. 2017, 8, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Sved Skottvoll, F.; Rayner, S.; Pedersen-Bjergaard, S.; Sullivan, G.; Krauss, S.; Ray Wilson, S.; Harrison, S. 3d cell culture models and organ-on-a-chip: Meet separation science and mass spectrometry. Electrophoresis 2020, 41, 56–64. [Google Scholar] [CrossRef]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef]
- Worley, K.E.; Rico-Varela, J.; Ho, D.; Wan, L.Q. Teratogen screening with human pluripotent stem cells. Integr. Biol. 2018, 10, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, L.; Guo, Y.; Zhu, Y.; Qin, J. Engineering stem cell-derived 3d brain organoids in a perfusable organ-on-a-chip system. RSC Adv. 2018, 8, 1677–1685. [Google Scholar] [CrossRef] [Green Version]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Chung, M.; Jeon, N.L. Engineering of functional, perfusable 3d microvascular networks on a chip. Lab Chip 2013, 13, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Paul, A.; Memic, A.; Khademhosseini, A. A multilayered microfluidic blood vessel-like structure. Biomed. Microdevices 2015, 17, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Phan, D.T.; Sobrino, A.; George, S.C.; Hughes, C.C.; Lee, A.P. Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels. Lab Chip 2016, 16, 282–290. [Google Scholar] [CrossRef]
- Chen, M.B.; Whisler, J.A.; Frose, J.; Yu, C.; Shin, Y.; Kamm, R.D. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 2017, 12, 865–880. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling development and the stem cell niche in a dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.W.; Schier, A.F. Morphogen gradients: From generation to interpretation. Annu. Rev. Cell Dev. Biol. 2011, 27, 377–407. [Google Scholar] [CrossRef] [Green Version]
- Demers, C.J.; Soundararajan, P.; Chennampally, P.; Cox, G.A.; Briscoe, J.; Collins, S.D.; Smith, R.L. Development-on-chip: In vitro neural tube patterning with a microfluidic device. Development 2016, 143, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.C.; Fernandes, T.G.; Diogo, M.M.; Cabral, J.M.S. Towards multi-organoid systems for drug screening applications. Bioengineering 2018, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, L.; Zhu, Y.; Qin, J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip 2018, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. A micro cell culture analog (microcca) with 3-d hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip 2009, 9, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic-pharmacodynamic (pk-pd) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef]
- Sung, J.H.; Srinivasan, B.; Esch, M.B.; McLamb, W.T.; Bernabini, C.; Shuler, M.L.; Hickman, J.J. Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med. 2014, 239, 1225–1239. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef]
- Mironov, V.; Boland, T.; Trusk, T.; Forgacs, G.; Markwald, R.R. Organ printing: Computer-aided jet-based 3d tissue engineering. Trends Biotechnol. 2003, 21, 157–161. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Y.; Liu, J. Bioprinting of 3d tissues/organs combined with microfluidics. RSC Adv. 2018, 8, 21712–21727. [Google Scholar] [CrossRef] [Green Version]
- Waheed, S.; Cabot, J.M.; Macdonald, N.P.; Lewis, T.; Guijt, R.M.; Paull, B.; Breadmore, M.C. 3D printed microfluidic devices: Enablers and barriers. Lab Chip 2016, 16, 1993–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdi, A.A.; Popma, A.; Wong, W.; Nguyen, T.; Pan, Y.; Xu, J. 3D printing: An emerging tool for novel microfluidics and lab-on-a-chip applications. Microfluid. Nanofluidics 2016, 20, 50. [Google Scholar] [CrossRef]
- Amin, R.; Knowlton, S.; Hart, A.; Yenilmez, B.; Ghaderinezhad, F.; Katebifar, S.; Messina, M.; Khademhosseini, A.; Tasoglu, S. 3D-printed microfluidic devices. Biofabrication 2016, 8, 22001. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Choudhury, D. Microfluidic bioprinting for organ-on-a-chip models. Drug Discov. Today 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Peng, W.; Datta, P.; Ayan, B.; Ozbolat, V.; Sosnoski, D.; Ozbolat, I.T. 3D bioprinting for drug discovery and development in pharmaceutics. Acta Biomater. 2017, 57, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, J.; Zhu, W.; Tang, M.; Lawrence, N.; Yu, C.; Gou, M.; Chen, S. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv. Drug Deliv. Rev. 2018, 132, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, J.; Varadaraj, S.; Dash, S.K.; Sharma, A.; Verma, R.S. Organotypic cancer tissue models for drug screening: 3D constructs, bioprinting and microfluidic chips. Drug Discov. Today 2020. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, G.; Musso, N.; Grasso, M.; Costantino, A.; Lazzarino, G.; Tascedda, F.; Gulisano, M.; Lunte, S.M.; Caraci, F. Microfluidics as a Novel Tool for Biological and Toxicological Assays in Drug Discovery Processes: Focus on Microchip Electrophoresis. Micromachines 2020, 11, 593. https://doi.org/10.3390/mi11060593
Caruso G, Musso N, Grasso M, Costantino A, Lazzarino G, Tascedda F, Gulisano M, Lunte SM, Caraci F. Microfluidics as a Novel Tool for Biological and Toxicological Assays in Drug Discovery Processes: Focus on Microchip Electrophoresis. Micromachines. 2020; 11(6):593. https://doi.org/10.3390/mi11060593
Chicago/Turabian StyleCaruso, Giuseppe, Nicolò Musso, Margherita Grasso, Angelita Costantino, Giuseppe Lazzarino, Fabio Tascedda, Massimo Gulisano, Susan M. Lunte, and Filippo Caraci. 2020. "Microfluidics as a Novel Tool for Biological and Toxicological Assays in Drug Discovery Processes: Focus on Microchip Electrophoresis" Micromachines 11, no. 6: 593. https://doi.org/10.3390/mi11060593