
Modeling Sarcoglycanopathy in Danio rerio
 , , , , , ,
, , , , , ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation and First Characterization of the δ-SG and β-SG KO Zebrafish Lines
2.2. Swimming Ability of sgcd−/− and sgcb−/− Larvae
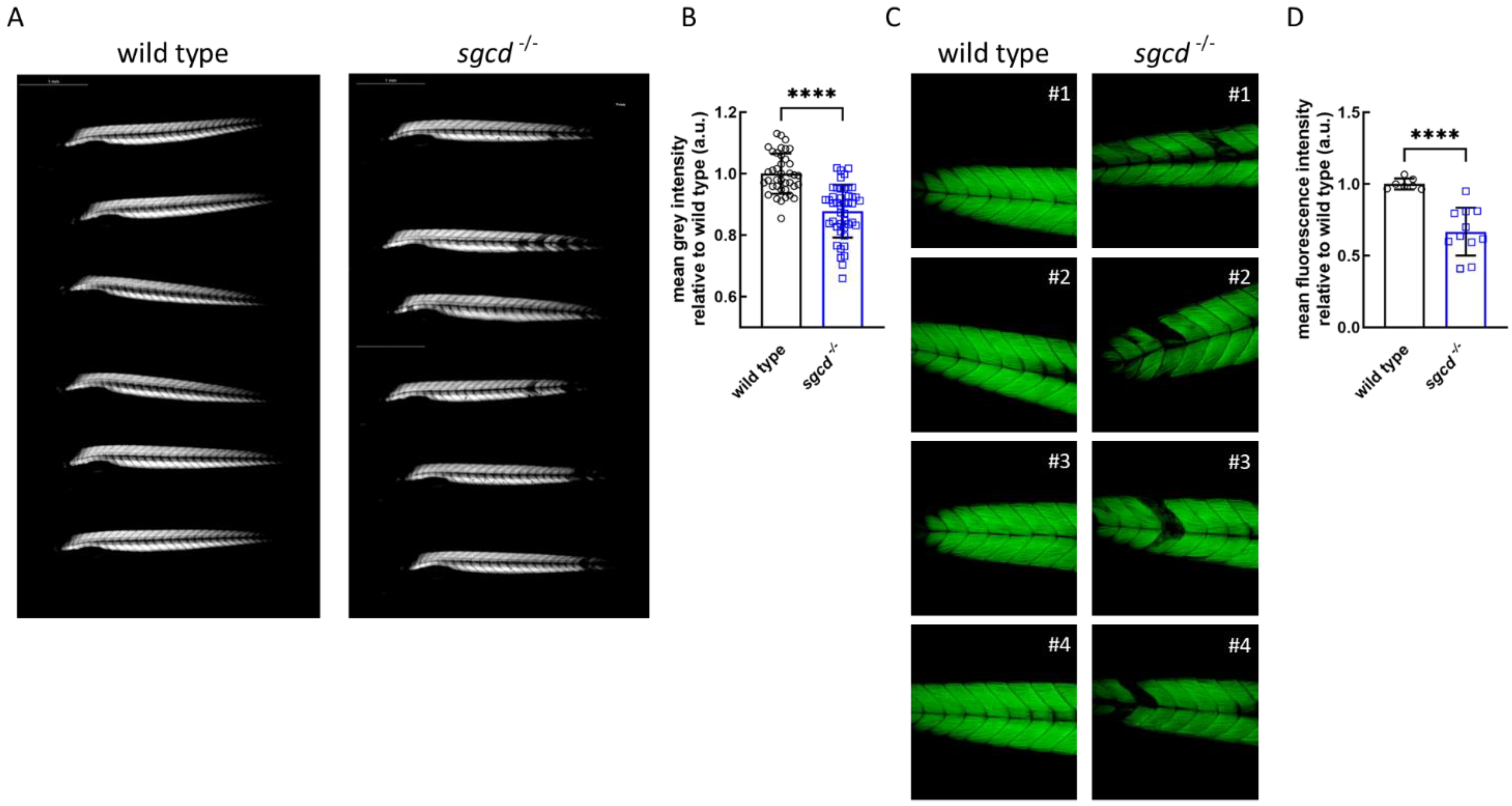
2.3. Analysis of Myofiber Integrity of sgcd−/− Larvae
2.4. Skeletal Muscle Structure and Ultrastructure of Wild Type and sgcd−/− Adult Fish
2.5. Swimming Performance of Adult sgcd−/−
2.6. Heart Phenotype of the Adult sgcd−/− Zebrafish
2.7. Skeletal Muscle Structure of Wild Type and sgcd−/− Larvae upon Growth in Stressful Condition
2.8. The E262K Missense Mutant of δ-SG Is Substrate of the ERAD Pathway in Zebrafish
3. Discussion
4. Material and Methods
4.1. Ethics Statement
4.2. Zebrafish Husbandry and Maintenance
4.3. Generation of Mutated Zebrafish Lines by CRISPR/Cas9 Genome Editing
4.4. Genomic DNA Extraction
4.5. Genotyping of sgcd and sgcb Mutants
4.6. Protein Extraction and Western Blot
4.7. Body Length Measurement
4.8. Phalloidin Staining
4.9. Morphological and Histological Analyses (H&E and Azan–Mallory)
4.10. Heart Dissection
4.11. Birefringence Assay
4.12. Larvae Locomotion Assays
4.13. Adult Fish Locomotion Assay
4.14. Swimming Tunnel Assay
4.15. Electron Microscopy
4.16. Re-Expression of Human δ-SG cDNA (Wild Type or Mutated) in sgcd−/− Embryos by mRNA Injection of Fertilized Oocytes
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainzof, M.; Souza, L.S.; Gurgel-Giannetti, J.; Zatz, M. Sarcoglycanopathies: An update. Neuromuscul. Disord. 2021, 31, 1021–1027. [Google Scholar] [CrossRef]
- Ozawa, E. Our trails and trials in the subsarcolemmal cytoskeleton network and muscular dystrophy researches in the dystrophin era. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 798–821. [Google Scholar] [CrossRef]
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci. (Landmark Ed.) 2016, 21, 744–756. [Google Scholar] [PubMed] [Green Version]
- Carotti, M.; Fecchio, C.; Sandona, D. Emerging therapeutic strategies for sarcoglycanopathy. Expert Opin. Orphan D 2017, 5, 381–396. [Google Scholar] [CrossRef]
- Angelini, C.; Fanin, M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Expert Opin Orphan D 2016, 4, 1239–1251. [Google Scholar] [CrossRef]
- Alonso-Perez, J.; Gonzalez-Quereda, L.; Bello, L.; Guglieri, M.; Straub, V.; Gallano, P.; Semplicini, C.; Pegoraro, E.; Zangaro, V.; Nascimento, A.; et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020, 143, 2696–2708. [Google Scholar] [CrossRef]
- Kirschner, J.; Lochmuller, H. Sarcoglycanopathies. Handb. Clin. Neurol 2011, 101, 41–46. [Google Scholar] [CrossRef]
- Politano, L.; Nigro, V.; Passamano, L.; Petretta, V.; Comi, L.I.; Papparella, S.; Nigro, G.; Rambaldi, P.F.; Raia, P.; Pini, A.; et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul. Disord. 2001, 11, 178–185. [Google Scholar] [CrossRef]
- Fanin, M.; Melacini, P.; Boito, C.; Pegoraro, E.; Angelini, C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul. Disord. 2003, 13, 303–309. [Google Scholar] [CrossRef]
- Ng, R.; Banks, G.B.; Hall, J.K.; Muir, L.A.; Ramos, J.N.; Wicki, J.; Odom, G.L.; Konieczny, P.; Seto, J.; Chamberlain, J.R.; et al. Animal Models of Muscular Dystrophy. Prog. Mol. Biol. Transl. 2012, 105, 83–111. [Google Scholar] [CrossRef] [Green Version]
- Gaina, G.; Popa Gruianu, A. Muscular dystrophy: Experimental animal models and therapeutic approaches (Review). Exp. Ther. Med. 2021, 21, 610. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M.; Cohn, R.D.; Hrstka, R.F.; Moore, S.A.; Allamand, V.; Davidson, B.L.; Williamson, R.A.; Campbell, K.P. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell 2000, 5, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Hack, A.A.; Cordier, L.; Shoturma, D.I.; Lam, M.Y.; Sweeney, H.L.; McNally, E.M. Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 10723–10728. [Google Scholar] [CrossRef]
- Coral-Vazquez, R.; Cohn, R.D.; Moore, S.A.; Hill, J.A.; Weiss, R.M.; Davisson, R.L.; Straub, V.; Barresi, R.; Bansal, D.; Hrstka, R.F.; et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: A novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999, 98, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Nigro, V.; Okazaki, Y.; Belsito, A.; Piluso, G.; Matsuda, Y.; Politano, L.; Nigro, G.; Ventura, C.; Abbondanza, C.; Molinari, A.M.; et al. Identification of the Syrian hamster cardiomyopathy gene. Hum. Mol. Genet. 1997, 6, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duclos, F.; Straub, V.; Moore, S.A.; Venzke, D.P.; Hrstka, R.F.; Crosbie, R.H.; Durbeej, M.; Lebakken, C.S.; Ettinger, A.J.; van der Meulen, J.; et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998, 142, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Zon, L.I.; Langenau, D.M. Zebrafish disease models in drug discovery: From preclinical modelling to clinical trials. Nat. Rev. Drug Discov. 2021, 20, 611–628. [Google Scholar] [CrossRef]
- Rosa, J.G.S.; Lima, C.; Lopes-Ferreira, M. Zebrafish Larvae Behavior Models as a Tool for Drug Screenings and Pre-Clinical Trials: A Review. Int. J. Mol. Sci. 2022, 23, 6647. [Google Scholar] [CrossRef] [PubMed]
- Steffen, L.S.; Guyon, J.R.; Vogel, E.D.; Beltre, R.; Pusack, T.J.; Zhou, Y.; Zon, L.I.; Kunkel, L.M. Zebrafish orthologs of human muscular dystrophy genes. BMC Genom. 2007, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medishetti, R.; Balamurugan, K.; Yadavalli, K.; Rani, R.; Sevilimedu, A.; Challa, A.K.; Parsa, K.; Chatti, K. CRISPR-Cas9-induced gene knockout in zebrafish. STAR Protoc. 2022, 3, 101779. [Google Scholar] [CrossRef] [PubMed]
- Hermsen, S.A.; van den Brandhof, E.J.; van der Ven, L.T.; Piersma, A.H. Relative embryotoxicity of two classes of chemicals in a modified zebrafish embryotoxicity test and comparison with their in vivo potencies. Toxicol. Vitr. 2011, 25, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Mosley, A.N.; Jun, S.J.; Montanaro, F.; Steffen, L.S.; Zhou, Y.; Nigro, V.; Zon, L.I.; Kunkel, L.M. Delta-sarcoglycan is required for early zebrafish muscle organization. Exp. Cell Res. 2005, 304, 105–115. [Google Scholar] [CrossRef]
- Cheng, L.; Guo, X.F.; Yang, X.Y.; Chong, M.; Cheng, J.; Li, G.; Gui, Y.H.; Lu, D.R. Delta-sarcoglycan is necessary for early heart and muscle development in zebrafish. Biochem. Biophys. Res. Commun. 2006, 344, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Best, C.; Kurrasch, D.M.; Vijayan, M.M. Maternal cortisol stimulates neurogenesis and affects larval behaviour in zebrafish. Sci. Rep. 2017, 7, 40905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rosa, V.; Frigato, E.; Lopez-Olmeda, J.F.; Sanchez-Vazquez, F.J.; Bertolucci, C. The Light Wavelength Affects the Ontogeny of Clock Gene Expression and Activity Rhythms in Zebrafish Larvae. PLoS ONE 2015, 10, e0132235. [Google Scholar] [CrossRef] [Green Version]
- Han, E.; Ho Oh, K.; Park, S.; Chan Rah, Y.; Park, H.C.; Koun, S.; Choi, J. Analysis of behavioral changes in zebrafish (Danio rerio) larvae caused by aminoglycoside-induced damage to the lateral line and muscles. Neurotoxicology 2020, 78, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Zarantoniello, M.; Randazzo, B.; Gioacchini, G.; Truzzi, C.; Giorgini, E.; Riolo, P.; Gioia, G.; Bertolucci, C.; Osimani, A.; Cardinaletti, G.; et al. Zebrafish (Danio rerio) physiological and behavioural responses to insect-based diets: A multidisciplinary approach. Sci. Rep. 2020, 10, 10648. [Google Scholar] [CrossRef] [PubMed]
- Morbiato, E.; Bilel, S.; Tirri, M.; Arfe, R.; Fantinati, A.; Savchuk, S.; Appolonova, S.; Frisoni, P.; Tagliaro, F.; Neri, M.; et al. Potential of the zebrafish model for the forensic toxicology screening of NPS: A comparative study of the effects of APINAC and methiopropamine on the behavior of zebrafish larvae and mice. Neurotoxicology 2020, 78, 36–46. [Google Scholar] [CrossRef]
- Lucon-Xiccato, T.; Tomain, M.; D’Aniello, S.; Bertolucci, C. bdnf loss affects activity, sociability, and anxiety-like behaviour in zebrafish. Behav. Brain Res. 2023, 436, 114115. [Google Scholar] [CrossRef]
- MacPhail, R.C.; Brooks, J.; Hunter, D.L.; Padnos, B.; Irons, T.D.; Padilla, S. Locomotion in larval zebrafish: Influence of time of day, lighting and ethanol. Neurotoxicology 2009, 30, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Beggs, A.H.; Gupta, V.A. Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J. Vis. Exp. 2013, 82, e50925. [Google Scholar] [CrossRef] [Green Version]
- Blain, A.M.; Straub, V.W. δ-Sarcoglycan-deficient muscular dystrophy: From discovery to therapeutic approaches. Skelet. Muscle 2011, 1, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Hou, Y.; Yu, M.; Liu, Y.; Fan, Y.; Zhang, W.; Wang, Z.; Xiong, H.; Yuan, Y. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J. Rare Dis. 2019, 14, 43. [Google Scholar] [CrossRef] [Green Version]
- Tesoriero, C.; Greco, F.; Cannone, E.; Ghirotto, F.; Facchinello, N.; Schiavone, M.; Vettori, A. Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays. Int. J. Mol. Sci. 2023, 24, 8314. [Google Scholar] [CrossRef]
- Lucon-Xiccato, T.; Bella, L.; Mainardi, E.; Baraldi, M.; Bottarelli, M.; Sandona, D.; Bertolucci, C. An Automated Low-Cost Swim Tunnel for Measuring Swimming Performance in Fish. Zebrafish 2021, 18, 231–234. [Google Scholar] [CrossRef]
- Wakamatsu, Y.; Ogino, K.; Hirata, H. Swimming capability of zebrafish is governed by water temperature, caudal fin length and genetic background. Sci. Rep. 2019, 9, 16307. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Sedmera, D.; Yost, H.J.; Clark, E.B. Structure and function of the developing zebrafish heart. Anat. Rec. 2000, 260, 148–157. [Google Scholar] [CrossRef]
- Hu, N.; Yost, H.J.; Clark, E.B. Cardiac morphology and blood pressure in the adult zebrafish. Anat. Rec. 2001, 264, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F. Cardiomyopathy in muscular dystrophies. Curr. Opin. Neurol. 2003, 16, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, A.A.; Oorschot, V.; Vaz, R.; Ramm, G.; Bryson-Richardson, R.J. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014, 128, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; D’Angelo, S.; Franzoso, S.; Fanin, M.; Angelini, C.; Betto, R.; Sandona, D. Inhibition of proteasome activity promotes the correct localization of disease-causing alpha-sarcoglycan mutants in HEK-293 cells constitutively expressing beta-, gamma-, and delta-sarcoglycan. Am. J. Pathol. 2008, 173, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, M.; Gicquel, E.; Barrault, L.; Soheili, T.; Malissen, M.; Malissen, B.; Vincent-Lacaze, N.; Perez, N.; Udd, B.; Danos, O.; et al. Mannosidase I inhibition rescues the human alpha-sarcoglycan R77C recurrent mutation. Hum. Mol. Genet. 2008, 17, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, E.; Fanin, M.; Mamchaoui, K.; Betto, R.; Sandona, D. Unveiling the degradative route of the V247M alpha-sarcoglycan mutant responsible for LGMD-2D. Hum. Mol. Genet. 2014, 23, 3746–3758. [Google Scholar] [CrossRef] [PubMed]
- Soheili, T.; Gicquel, E.; Poupiot, J.; N’Guyen, L.; Le Roy, F.; Bartoli, M.; Richard, I. Rescue of sarcoglycan mutations by inhibition of endoplasmic reticulum quality control is associated with minimal structural modifications. Hum. Mutat. 2012, 33, 429–439. [Google Scholar] [CrossRef]
- Winder, S.J.; Lipscomb, L.; Angela Parkin, C.; Juusola, M. The proteasomal inhibitor MG132 prevents muscular dystrophy in zebrafish. PLoS Curr. 2011, 3, RRN1286. [Google Scholar] [CrossRef] [PubMed]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Zulian, A.; Schiavone, M.; Giorgio, V.; Bernardi, P. Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol. Res. 2016, 113 Pt A, 563–573. [Google Scholar] [CrossRef]
- Vainzof, M.; Ayub-Guerrieri, D.; Onofre, P.C.; Martins, P.C.; Lopes, V.F.; Zilberztajn, D.; Maia, L.S.; Sell, K.; Yamamoto, L.U. Animal models for genetic neuromuscular diseases. J. Mol. Neurosci. 2008, 34, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Ono, K.; Abe, M.; Jasmin, G.; Eki, T.; Murakami, Y.; Masaki, T.; Toyo-oka, T.; Hanaoka, F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: An animal model of disrupted dystrophin-associated glycoprotein complex. Proc. Natl. Acad. Sci. USA 1997, 94, 13873–13878. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Saito, N.; Watano, K.; Igarashi, K.; Tagami, S.; Shima, H.; Kikuchi, K. Defect of delta-sarcoglycan gene is responsible for development of dilated cardiomyopathy of a novel hamster strain, J2N-k: Calcineurin/PP2B activity in the heart of J2N-k hamster. J. Biochem. 2003, 134, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.F.; Patissier, C.; Bourg, N.; Fecchio, C.; Sandona, D.; Marsolier, J.; Richard, I. Different outcome of sarcoglycan missense mutation between human and mouse. PLoS ONE 2018, 13, e0191274. [Google Scholar] [CrossRef] [Green Version]
- Kobuke, K.; Piccolo, F.; Garringer, K.W.; Moore, S.A.; Sweezer, E.; Yang, B.; Campbell, K.P. A common disease-associated missense mutation in alpha-sarcoglycan fails to cause muscular dystrophy in mice. Hum. Mol. Genet. 2008, 17, 1201–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jia, Z.; Zhang, S.; He, X. Progress in Gene-Editing Technology of Zebrafish. Biomolecules 2021, 11, 1300. [Google Scholar] [CrossRef]
- Burgess, H.A.; Granato, M. Modulation of locomotor activity in larval zebrafish during light adaptation. J. Exp. Biol. 2007, 210, 2526–2539. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125 Pt B, 122–131. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Gunther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Joris, M.; Schloesser, M.; Baurain, D.; Hanikenne, M.; Muller, M.; Motte, P. Number of inadvertent RNA targets for morpholino knockdown in Danio rerio is largely underestimated: Evidence from the study of Ser/Arg-rich splicing factors. Nucleic Acids Res. 2017, 45, 9547–9557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krylov, V.V.; Izvekov, E.I.; Pavlova, V.V.; Pankova, N.A.; Osipova, E.A. Circadian rhythms in zebrafish (Danio rerio) behaviour and the sources of their variability. Biol. Rev. Camb. Philos. Soc. 2021, 96, 785–797. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front. Cell Dev. Biol. 2022, 10, 952249. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Al-Qusairi, L.; Laporte, J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 2011, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial stress responses in Duchenne muscular dystrophy: Metabolic dysfunction or adaptive reprogramming? Am. J. Physiol. Cell Physiol. 2022, 323, C718–C730. [Google Scholar] [CrossRef]
- Schade van Westrum, S.M.; Dekker, L.R.; de Voogt, W.G.; Wilde, A.A.; Ginjaar, I.B.; de Visser, M.; van der Kooi, A.J. Cardiac involvement in Dutch patients with sarcoglycanopathy: A cross-sectional cohort and follow-up study. Muscle Nerve 2014, 50, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Widrick, J.J.; Alexander, M.S.; Sanchez, B.; Gibbs, D.E.; Kawahara, G.; Beggs, A.H.; Kunkel, L.M. Muscle dysfunction in a zebrafish model of Duchenne muscular dystrophy. Physiol. Genom. 2016, 48, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farr, G.H., 3rd; Morris, M.; Gomez, A.; Pham, T.; Kilroy, E.; Parker, E.U.; Said, S.; Henry, C.; Maves, L. A novel chemical-combination screen in zebrafish identifies epigenetic small molecule candidates for the treatment of Duchenne muscular dystrophy. Skelet. Muscle 2020, 10, 29. [Google Scholar] [CrossRef]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef]
- Dang, M.; Henderson, R.E.; Garraway, L.A.; Zon, L.I. Long-term drug administration in the adult zebrafish using oral gavage for cancer preclinical studies. Dis. Model Mech. 2016, 9, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Kinkel, M.D.; Eames, S.C.; Philipson, L.H.; Prince, V.E. Intraperitoneal injection into adult zebrafish. J. Vis. Exp. 2010, 30, e2126. [Google Scholar] [CrossRef]
- Pugach, E.K.; Li, P.; White, R.; Zon, L. Retro-orbital injection in adult zebrafish. J. Vis. Exp. 2009, 34, e1645. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.T.; Doerr, K.M.; Whipps, C.M. Antibiotic treatment of zebrafish mycobacteriosis: Tolerance and efficacy of treatments with tigecycline and clarithromycin. J. Fish Dis. 2017, 40, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Patton, E.E. Long-term non-invasive drug treatments in adult zebrafish that lead to melanoma drug resistance. Dis. Model Mech. 2022, 15, dmm049401. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Kunkel, L.M. Zebrafish based small molecule screens for novel DMD drugs. Drug Discov. Today Technol. 2013, 10, e91–e96. [Google Scholar] [CrossRef] [PubMed]
- Hyzewicz, J.; Ruegg, U.T.; Takeda, S. Comparison of Experimental Protocols of Physical Exercise for mdx Mice and Duchenne Muscular Dystrophy Patients. J. Neuromuscul. Dis. 2015, 2, 325–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, A.; Carra, S.; De Blasio, P.; Cotelli, F.; Biunno, I. Sel1l knockdown negatively influences zebrafish embryos endothelium. J. Cell Physiol. 2018, 233, 5396–5404. [Google Scholar] [CrossRef]
- Chen, Z.; Ballar, P.; Fu, Y.; Luo, J.; Du, S.; Fang, S. The E3 ubiquitin ligase gp78 protects against ER stress in zebrafish liver. J. Genet. Genom. 2014, 41, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Imamura, S.; Yabu, T.; Yamashita, M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J. Biol. Chem. 2012, 287, 23047–23056. [Google Scholar] [CrossRef] [Green Version]
- Imai, F.; Yoshizawa, A.; Fujimori-Tonou, N.; Kawakami, K.; Masai, I. The ubiquitin proteasome system is required for cell proliferation of the lens epithelium and for differentiation of lens fiber cells in zebrafish. Development 2010, 137, 3257–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fu, X.; Gaiser, S.; Kottgen, M.; Kramer-Zucker, A.; Walz, G.; Wegierski, T. OS-9 regulates the transit and polyubiquitination of TRPV4 in the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 36561–36570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, P. Calcium Imaging in the Zebrafish. In Calcium Signaling; Advances in Experimental Medicine and Biology; Islam, M., Ed.; Springer: Cham, Switzerland, 2020; Volume 1131. [Google Scholar]
- Salgado-Almario, J.; Vicente, M.; Molina, Y.; Martinez-Sielva, A.; Vincent, P.; Domingo, B.; Llopis, J. Simultaneous imaging of calcium and contraction in the beating heart of zebrafish larvae. Theranostics 2022, 12, 1012–1029. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalla Barba, F.; Soardi, M.; Mouhib, L.; Risato, G.; Akyürek, E.E.; Lucon-Xiccato, T.; Scano, M.; Benetollo, A.; Sacchetto, R.; Richard, I.; et al. Modeling Sarcoglycanopathy in Danio rerio. Int. J. Mol. Sci. 2023, 24, 12707. https://doi.org/10.3390/ijms241612707
Dalla Barba F, Soardi M, Mouhib L, Risato G, Akyürek EE, Lucon-Xiccato T, Scano M, Benetollo A, Sacchetto R, Richard I, et al. Modeling Sarcoglycanopathy in Danio rerio. International Journal of Molecular Sciences. 2023; 24(16):12707. https://doi.org/10.3390/ijms241612707
Chicago/Turabian StyleDalla Barba, Francesco, Michela Soardi, Leila Mouhib, Giovanni Risato, Eylem Emek Akyürek, Tyrone Lucon-Xiccato, Martina Scano, Alberto Benetollo, Roberta Sacchetto, Isabelle Richard, and et al. 2023. "Modeling Sarcoglycanopathy in Danio rerio" International Journal of Molecular Sciences 24, no. 16: 12707. https://doi.org/10.3390/ijms241612707