1. Introduction
Ascochyta Blight (AB), also referred to as black spot, is a devastating disease of many cool-season legumes of commercial importance, including field pea, lentil, chickpea, and faba bean. AB, caused by fungal species from the phylum Ascomycota, is a foliar disease affecting all of the above-ground organs of the plant, including the stem, leaves, pods, and flowers. AB affects most legume growing regions around the world, especially Europe, Australia, New Zealand, and North America. It is a major production constraint in all of the areas where these crops are produced, presenting significant crop losses as well as a reduction in yield and grain quality [
1,
2,
3].
Unlike other legumes where only one ascochyta species is responsible for the disease, there are at least seven reported species which are responsible for AB in field peas, including:
Ascochyta pisi Lib.,
Peyronellaea pinodes (Berk. and A. Bloxham) (Synonyms:
Ascochyta pinodes, Mycosphaerella pinodes, Teleomorph:
Didymella pinodes),
Peyronellaea pinodella (L.K. Jones) (Synonym:
Ascochyta pinodella, syn.
Phoma medicaginis var. pinodella),
Phoma koolunga, Phoma herbarum, Boerma exigua var. exigua, and
Phoma glomerata [
4,
5,
6,
7,
8,
9,
10]. Significant yield losses have been reported across the world, although varying environmental conditions and cropping systems affect the level of damage due to ascochyta blight. Up to 50% losses have been reported in Canada, 40% in France, 53% in Ethiopia, and 30% in China; a disease severity of 72% has been reported in Spain [
11,
12,
13,
14,
15]. In Australia, AB is the second most important biotic stress affecting field pea production after bacterial blight caused by
Pseudomonas syringae. Average annual losses to AB are from 10 to 60% in Australia, with higher losses observed when environmental conditions such as rain and wind favour disease progression [
16,
17]. Economic losses can occur through a reduction in crop biomass or the infection of pods and seeds that makes the grain unmarketable [
18,
19,
20,
21,
22].
In Australia, AB presents as a complex of mainly
P. pinodes;
P. pinodella and
P. koolunga in varying proportions, with
P. pinodella seemingly being the least prevalent. With three causal pathogen species, the management of AB is challenging and complex, and requires a combination of management strategies [
16,
20,
23,
24]. The frequency of individual pathogen species varies within the growing season, further limiting the effectiveness of more generalized management approaches. The most efficient management system is the use of resistant cultivars; however, due to the multi-species disease complex, breeding for AB resistance has been difficult [
16,
25]. Resistance to one species has no significant impact on crop and yield loss because this leads to the increased prevalence of the other AB complex species infecting the crop.
The characterization of AB species in chickpea and lentil is well advanced [
26,
27,
28]; nonetheless, there is a gap in the knowledge of the AB pathogen complex in field pea. The morphological characterization of AB species can vary significantly, and is largely dependent on culturing techniques; the similar colony morphology for different species also makes it challenging to assign isolates to species [
29]. Extensive genetic characterization of the AB complex is lacking, and in addition,
P. pinodes and
P. pinodella are not easily distinguishable using current molecular methods due to their high genomic similarity.
P. koolunga, discovered in 2009, seems to be more taxonomically different from
P. pinodes and
P. pinodella, [
29,
30,
31,
32]. Whole-genome assemblies are required in order to characterize the different AB species, understand host–pathogen interactions, and enable genomically assisted breeding for the development of resistant varieties. To date, no whole genome sequence assembly of the field pea Ascochyta species has been published, although there is a published mitochondrial genome for
M. pinodes [
33].
Whole-genome information is vital for the development of reliable field diagnostic tests that can accurately identify different AB species that can co-exist on the crop. This will advance our understanding of the disease epidemiology [
6,
29] and help to define appropriate management strategies. Research reveals that sexual reproduction and host genetic diversity contribute to virulence; therefore, the determination of the mating type for the AB species—especially for
P. koolunga, the mating type of which has not yet been determined—could drive further research [
16,
34]. Understanding host–pathogen interactions is also crucial for the elucidation of the primary features of the plant immune response. These findings can unravel the genetic mechanisms underlying plant defenses and disease resistance, which facilitates functional marker development and application in crop breeding programs. Fungal whole-genome assemblies can help with the eludication of host–pathogen interactions because fungal genome plasticity and transposable elements enable them to evolve and adapt at a rapid rate, making it challenging to develop suitable management strategies to aid plant protection [
35,
36,
37]. Thus, an in-depth genetic characterization of the main AB complex species is an important first step towards finding better ways to control disease outbreaks, develop targeted and effective fungicides, and advance the development of AB-resistant field pea varieties.
One approach to better understand the genetic diversity and gene distribution within species is to combine comparative genomics with pan-genome analysis. Comparative genomics compares the genomic content of one species to another at micro- as well as macro-syntenic levels, based on gene annotation, the characterization of genome plasticity, and other evolutionary events, and resolves phylogenetic relationships [
38,
39,
40,
41]. While comparative genomic analysis can span kingdoms, pan-genome analysis was developed to narrow down the understanding of similarities and differences between strains or isolates of the same species or genus [
42,
43,
44,
45,
46]. Pan-genome analysis enables the investigation of the functional diversity and abundance of genes, as well as the core and accessory gene distribution between species. Genome-based studies have shown that lineage-specific accessory genes can encode virulence determinants (avirulence genes), and the identification of such genes can enable targeted breeding for the improvement of plant resistance genes in order to prevent disease [
47,
48,
49,
50].
Advances in next-generation sequencing technologies (NGS) have enabled the sequencing of many non-model organisms at a reduced cost. This provides genetic resources that are useful for comprehensive comparative analysis. NGS includes short-read sequencing technologies such as Illumina and long-read sequencing platforms such as Oxford Nanopore Technologies (ONT) [
51,
52,
53]. The assembly methods for short-read sequencing lack the resolution and capability to account for structural variants and sequence repeats, which are major features of complex eukaryotes such as fungi. Biodiversity research using molecular markers that exceed 550 bp for greater taxonomic resolution is also significantly impeded [
54,
55,
56,
57]. In contrast, long-read sequencing technologies resolves all of these drawbacks through individual reads long enough to cover entire repeat sequences. and the bioinformatic tools available for error correction and polishing can help to resolve their high error rate [
58,
59,
60,
61,
62,
63,
64]. Consequently, species and genotype-level taxonomic resolutions have been greatly improved using long-read sequencing in fungi, which in turn, could improve disease monitoring strategies, epidemiological studies and diagnostic tool development [
65,
66].
To the best of our knowledge, no genome assembly is available for these three AB species to date. Using long- and short-read sequencing technology, the aim of this study was to obtain high-quality genome assemblies that will play an important role in future research to understand these species and how they interact with their host plant and the environment. Here, we present the first fully annotated nuclear and mitochondrial draft genome assemblies for 18 AB isolates, six each of P. pinodes, P. pinodella and P. koolunga. Our investigations included comparative and pan-genome analysis, genome annotation, and the exploration of secondary metabolites, CAZymes, orthologs and phylogenetic relationships. We have generated a valuable resource.
2. Materials and Methods
2.1. Fungal Culture
Eighteen field pea AB isolates were obtained from the Ascochyta pathogen culture collection at the Centre for Crop and Disease Management, Curtin University, Western Australia. The isolates were received as dried cultures on filter paper preserved with desiccants in airlocked bags. Six isolates for each species were used for this study. These included P. pinodes (Isolate3Pp; Isolate4Pp; Isolate5Pp; Isolate87Pp; Isolate88Pp; Isolate97Pp), P. pinodella (Isolate18Ppll; Isolate27Ppll; Isolate72Ppll; Isolate58Ppll; Isolate104Ppll; Isolate113Ppll) and P. koolunga (Isolate1Pk; Isolate2Pk; Isolate22Pk; Isolate32Pk; Isolate36Pk; Isolate42Pk). The isolates were cultured on potato dextrose agar (Difco laboratories, USA) and grown in a temperature- and light-controlled incubator for 5–15 days with a photoperiod of 12 h/23 °C dark and 12 h/19 °C black light (a UV-A fluorescent tube). The mycelial plugs were cut from media plates and grown in potato dextrose broth for 3–4 days at 22 °C in an incubator shaker at 150 rpm in the dark.
2.2. Nucleic Acid Extraction
For the DNA extraction, mycelial samples were harvested and freeze dried for 24 h. The total DNA was isolated from the freeze-dried samples using the Wizard® Genomic DNA Purification System (Promega, Madison, WI, USA) as per the manufacturer’s instructions to achieve high-molecular-weight DNA. The eluted DNA was quantified using a QubitTM dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), and the quality was assessed using a Nanodrop 2000 (Thermo Fisher Scientific). The integrity and molecular weight were assessed using an Agilent TapeStation 2200 system (Agilent Technologies, Santa Clara, CA, USA) following manufacturer’s instructions.
For RNA extraction, mycelial samples were collected into 1.5 mL Eppendorf tubes and stored at −80 °C until they were processed. The RNA extraction was carried out using a Plant/Fungi Total RNA Purification Kit (Norgen Biotek Corp., Thorold, ON, Canada). Quality was assessed using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific), and integrity was assessed using an Agilent TapeStation 2200 system.
2.3. Oxford Nanopore Technologies (ONT) Sequencing
All of the ONT flow cells, library preparation kits and other consumables were sourced from Oxford Nanopore Technology (Oxford, England). The genome libraries for the isolates P. pinodes (Isolate3Pp; Isolate4Pp; Isolate5Pp), P. pinodella (Isolate18Ppll) and P. koolunga (Isolate1Pk; Isolate2Pk) were prepared using the ONT 1D ligation sequencing kit (SQK-LSK109) for MinION, with no shearing. For the remaining isolates, genome libraries were prepared using the SQK-LSK109 native barcode genomic DNA protocol for PromethION. The libraries were prepared according to the manufacturer’s instructions; however, the loading concentrations were adjusted to achieve the optimum loading of the flow cell.
Six high-molecular-weight ONT libraries with sizes ranging from 20 Kb to approximately 60 Kb for Isolate1Pk, Isolate2Pk, Isolate3Pp, Isolate4Pp, Isolate5Pp and Isolate18Ppll were sequenced using the ONT MinION platform. Each library was sequenced on one R9.4 flow cell (FLO-MIN106, ONT), except for Isolate2Pk, which was sequenced on two R9.4 flow cells, and the reads were combined. Additional sequencing was undertaken with all 18 high-molecular-weight ONT libraries, with sample sizes ranging from 10 Kb to approximately 60 Kb being sequenced using the ONT PromethION platform. Each library was sequenced on a PromethIon flow cell (FLO-PRO002, ONT). Fastq data from MinION and PromethION for each isolate were combined before proceeding to further analysis.
2.4. Transcriptome Sequencing and Alignment
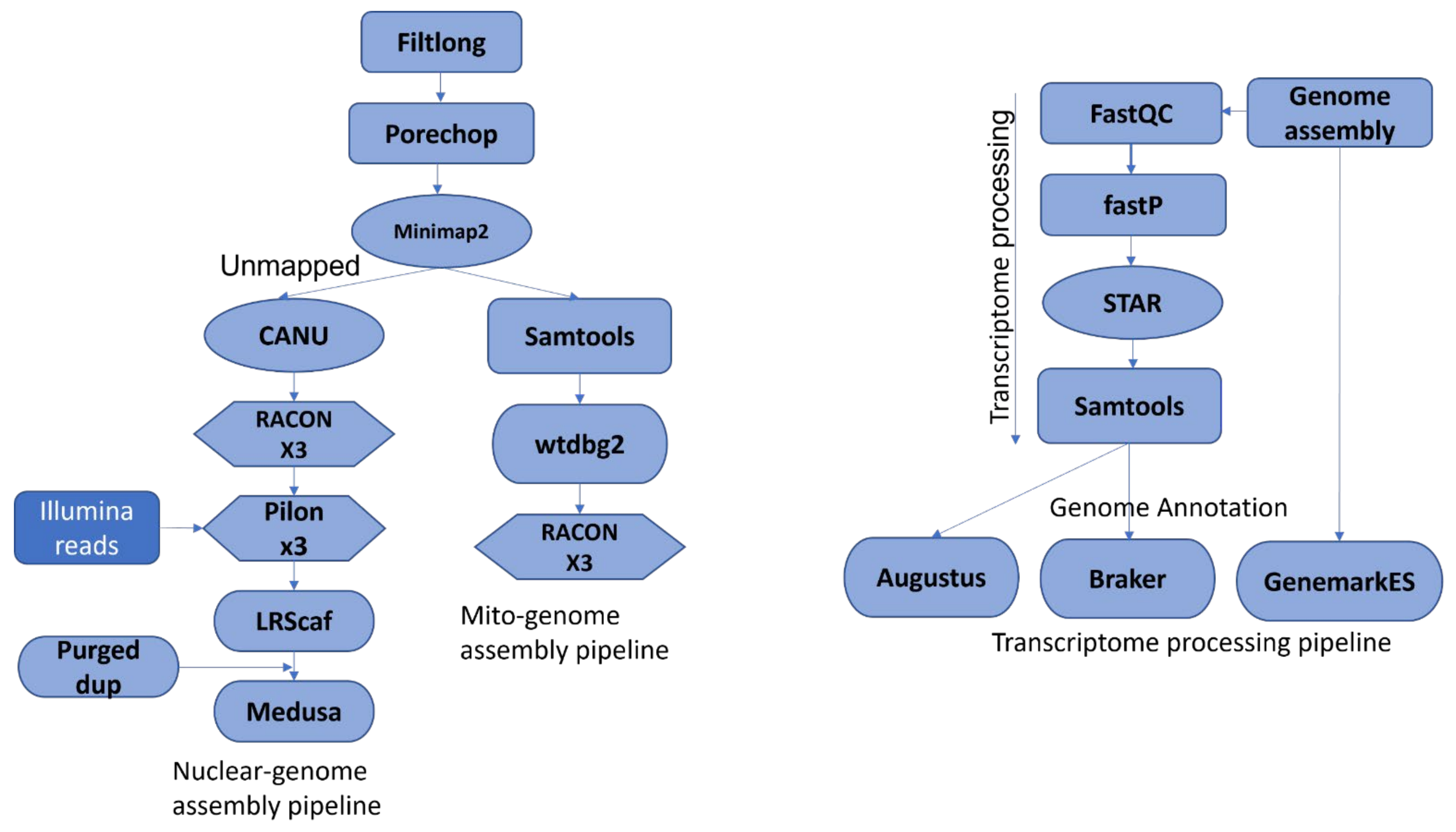
RNA samples extracted as detailed above were used to prepare transcriptome libraries for all 18 isolates in triplicate using Agilent’s Sure Select Strand Specific RNA Library Preparation Kit (Agilent Technologies) according to the manufacturer’s protocol. The RNA concentrations were estimated by running a sample of each library on an Illumina MiSeq sequencer (Illumina, Inc., San Diego, CA, USA) to measure library-specific barcode concentrations, and were pooled to achieve even proportions of each library in the pooled sample. Paired end sequencing (151 cycles + 151 cycles) was carried out on a NovaSeq 3000 (Illumina, Inc., San Diego, CA, USA). The RNA-seq data were processed according to the pipeline shown in
Figure 1. FastQC v0.11.8 (
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ was used to check the quality of the sequencing data. Fastp v0.21.0 [
67] was used to trim the adaptors. STAR v2.7.3 [
68] was used to align the processed transcripts to the respective genomes generating transcript alignment files in BAM format to be used for genome annotation.
2.5. Repeat Expression Analysis
The results from RepeatMasker for all of the isolates in GFF3 format were used to extract repeat sequences from the assembled AB genomes with getfasta in bedtools [
69]. Repeat fasta files were separated by species and concatenated into a single file per species before clustering to obtain only the unique repeats present in each species using CDHITS [
70]. The transcripts were quantified using Salmon [
71] to obtain the number of reads for each repeat element.
2.6. Nuclear and Mitochondrial Genome Assembly, and Post Assembly Analysis
Sequencing data from MinION and PromethION was acquired using MinKNOW v0.51.1.62 (Oxford Nanopore Technologies. Oxford, United Kingdom). Raw fastq reads were processed as shown in
Figure 1 and were categorized into “pass” or “fail” based on an average quality score > Q7. Only fastq pass reads were used for further processing and genome assembly. The reads were first filtered based on quality using the Filtlong package v0.2.0 (
https://github.com/rrwick/Filtlong), and then adaptors were removed using the Porechop package v0.2.4 (
https://github.com/rrwick/Porechop. Minimap2 v10.2.0 [
72] was used to extract mitochondrial reads through the sequence alignment of processed reads to the published
M. pinodes mitochondrial genome [
73]. The post-processing of sequence alignment data, including conversion from SAM to BAM format and the sorting of BAM files, before separating mapped and unmapped reads as fastq files, was carried out using Samtools v1.12 [
74]. Mapped reads were processed to assemble mitochondrial genomes, and unmapped reads were processed to generate nuclear genome assemblies for each isolate. Nuclear genomes were assembled with CANU v2.0 [
75]. Approximate genome sizes for all three species were determined using CANU, wtdgb2 v2.2, [
76] and Flye v2.6 [
77] for input into CANU:
P. pinodes (35 Mbp),
P. pinodella (45 Mbp),
P. koolunga (60 Mbp). Assemblies were carried out with three different minReadLength values (1000, 3000, 5000), and the assemblies with the lowest number of contigs were chosen as the final assemblies for each isolate. These draft assemblies were polished three times using Racon v1.4.10 [
78] and Pilon v1.23 [
79]. The nuclear assemblies for Isolate1Pk, Isolate2Pk, Isolate3Pp, Isolate4Pp, Isolate5Pp had Illumina short-reads available on the SRA database (SRR8284140, SRR8284141, SRR8284142, SRR8284136, SRR8284138, SRR8284139), and these were used for the hybrid Pilon polishing. The assembled genomes were assessed for quality using Quast v5.0.2 [
80], and scaffolding was carried out using LRScaf v1.1.10 [
81] and MeDuSa v1.6 [
82]. The completeness of the genome assemblies was determined using BUSCO [
83], and duplicated genes were removed using PurgeDup v20201029 [
84]. Final genome assemblies were again assessed using the BUSCO Fungi_Odb10 database on GenSAS (JL Humann, et al., 2019).
Known fungal single-copy genes, elongation factor 1-α (EF1-α), elongation factor 3-α (EF3-α), and the second largest subunit of RNA polymerase II (RPB2) were used to test for duplications in the assemblies that would indicate assembly errors using Blastn. The sequences used included Aspergillus tanneri translational elongation factor EF-1α (TEF3; XM_033570262.1), Ascochyta pisi strain CBS 122785 DNA-directed RNA polymerase II second-largest subunit (RPB2; MT018244.1), and Aspergillus oryzae strain translation elongation factor 1- α (tef-1-α; JCM10114).
The mitochondrial genomes were assembled using wtdgb2. The assemblies were quality checked by increasing the value of the estimated genome size parameter -g (65, 70, 80) in order to confirm the mitochondrial genome size and contig number before polishing three times using Racon version 1.4.10.
2.7. Genome Annotation
Automated genome annotation was carried out using the GenSAS web server platform, which includes pipelines and databases developed for fungi accessed on 11 Jan 2021. The transcriptome data generated above were used for the further refinement of the gene annotation. Detailed annotation of repeats in the genome assemblies was carried out using RepeatMasker v4.0.7 [
85] and RepeatModeler v1.0.11 (
http://www.repeatmasker.Org. RIPper [
86], a web-based tool, was used to identify repeat-induced mutation activity in the assembled genomes of the reference isolates for each of the three AB species. Repeats were masked for all of the assemblies before the functional and structural annotation was performed. Transcripts were predicted and aligned using BLAST nucleotide (blastn) [
87], BLAT [
88], and Program to Assemble Spliced Alignments (PASA) [
89] against NCBI refseq fungi databases with default parameters. Trembl, SwissProt (
https://www.uniprot.org/) and DIAMOND proteins v0.9.22 [
90] were used to predict the protein sequence with NCBI refseq fungi and database default parameters. Gene prediction was performed using Augustus v3.3.1 [
91] with the
A. fumigatus library as a hint annotation model, GenemarkES v4.38 [
92], and Braker v2.1.1 (
https://github.com/Gaius-Augustus/BRAKER). The most complete prediction was used for further downstream annotation. RNAmmer tool v1.2 [
93] was used to annotate 5s/8s, 16s/18s, and 23s/28s ribosomal RNA in the nuclear genomic sequences. SSR Finder v1.0 (
http://www.fresnostate.edu/ssrfinder/) was used to find Simple Sequence Repeats (SSR), and tRNAscan-SE v2.0 [
94] was used to identify tRNA genes in the AB genome assemblies. Functional annotation was carried out to enable the assignment of function and gene names. Conserved protein domains for the gene models were annotated using BLAST protein vs protein (blastp) version 2.7.1, DIAMOND, InterProScan v5.29–68.0 [
95], and PFAM v1.6 [
96]. SignalP v4.1 [
97] was used to predict the presence and location of signal peptide cleavage sites in the predicted protein sequences. The completeness and quality of the structural annotation was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) v3 with the Fungi_Odb10 database. GC content analysis was carried out using a 1000k sliding window.
2.8. AB GC Content Variation
Each genome was cut into non-overlapping 1 kb pieces, and the GC content of each 1 kb piece was measured. The %GC for each piece was rounded to an integer value and summed by the %GC value for each genome to create a %GC frequency distribution.
2.9. AB Nuclear Genome Structural Variation and Synteny Analysis
Genome-wide comparisons of structural variation between pairs of AB reference genome assemblies were carried out using Nucmer v4.0 [
99] to generate delta alignment files that were used as input into Assemblytics [
100]. Genome similarity data were generated for reference isolates (Isolate2Pk
P. koolunga, Isolate3Pp
P. pinodes and Isolate18Ppll
P. pinodella) using Nucmer with the --mum setting. The identification and analysis of the genome similarities among AB isolates was carried out using CIRCOS software v 0.69–9 [
101] using the links function to illustrate genomic regions identified by Nucmer to have a high level of nucleotide sequence homology.
2.10. BLASTn Similarity and Blast2go Analysis
Sequence similarity checks for the assembled nuclear genomes against the NCBI fungal genome nr/nt database were carried out using blastn, blastp and blastx. The parameters were set to allow only the top hits for each contig in the assembly being queried. Blast2go software was used to generate the top blast hits for the genome assemblies using annotated CDS gene files for reference isolates within each AB species.
2.11. Mating Type Determination
A. lentis MAT1-1 (DQ341314.1) and MAT1-2 (DQ341315.2), as well as P. pinodella MAT1-1 (JF815529.1) and MAT1-2 (JF815531.1) genes (accessed on 25 February 2021), were blasted against assembled nuclear genomes using blastn to determine their mating type according to homologous genes described by Cherif et al. (2006).
2.12. Ortholog Analysis
Predicted protein sequences in FASTA format for the AB species were used to predict orthologous gene clusters with the web program OrthoVenn [
102], which utilizes OrthoMCL, a well-known heuristic approach for the identification of ortholog groups [
103]. Gene families were analysed, and core and accessory genes were identified and used to describe pangenomes for the respective field pea AB species.
2.13. Phylogenetic Analysis
Whole-genome phylogeny for the field pea AB isolates was determined using kSNP3 [
104]. The resulting parsimony tree was uploaded into FigTree version 1.4.3 (
http://tree.bio.ed.ac.uk/software/figtree/) for drawing, rooting, and ordering nodes. OrthoFinder version 2.3.8 [
105] was also used to infer phylogeny using predicted protein sequences. Fourteen fungal species sourced on 4 February 2021 from public databases (NCBI) were included in the analysis, in order to determine the phylogenetic relationships. All of the species used in the analysis are from the phylum Ascomycota, except
Ustilago maydis, which is from the phylum Basidiomycota. Using Nei’s pairwise genetic distance calculation method, neighbour-joining (NJ) dendrograms were generated for rDNA internal transcribed spacer (ITS) and protein coding genes β-tubulin, the second largest subunit of RNA polymerase II (RPB2), transcription elongation factor 1-α (TEF1α) and topoisomerase 1 (TOP1), with NCBI accession numbers GU237883.1, KX838493.1, AB007770.1, and NW_017306212.1, respectively (accessed on 11 February 2021).
2.14. Carbohydrate-Active Enzyme (CAZymes) Analysis
CAZymes annotation was carried out using the dbCAN2 annotation pipeline (
http://bcb.unl.edu/dbCAN2/, accessed on 26 August 2021), a web-based application [
106]. This application annotates for putative homology to protein sequences in the CAZymes database using three tools: (i) HMMER, a dbCAN HMM (hidden Markov model) database [
107]; (ii) DIAMOND, a CAZy pre-annotated CAZyme sequence database [
90]; and (iii) Hotpep, a conserved CAZyme short peptide database [
108].
2.15. Secondary Metabolite Gene Cluster Analysis
5. Discussion
Although AB of field peas has been studied for many years, with literature on the subject dating as far back as 1944 [
112], there remain significant gaps in the literature that limit our understanding of the essential features of the disease. Elements such as the molecular basis underlying the pathogenesis, pathogen–complex interaction, host–pathogen interaction, and disease epidemiology require the detailed genome analysis of the pathogens causing AB. This study reports the first whole-genome assemblies and comparative analysis of
P.
koolunga,
P. pinodella and
P. pinodes, the three primary species of the AB complex in Australia. The draft genome assemblies generated from long-read sequencing were within the expected genome size for the Ascomycota phylum (average 36.9 Mb), with some isolates having genomes larger than the average genome size for plant pathogenic fungi of 39.4 Mb [
113,
114,
115]. The genome sizes for
P. pinodella and
P. pinodes at approximately 36 Mb were comparable to the assembled draft genomes of other Ascochyta species affecting legumes such as
A. rabiei (40 Mb) [
116] and
A. lentis (42 Mb) [
117]. However, a larger genome of approximately 56 Mb was observed for
P. koolunga, which is unsurprising, as large genomes have been confirmed for Dothideomycete species [
118,
119,
120]. The large genome size for
P. koolunga is almost entirely due to a high proportion of repeat elements, including repeat expansion and contraction, and this could be the driving force for genome evolution in the species, as has been observed for
Pyrenophora teres that cause net blotch in barley, with genome sizes ranging from 41 Mb to 51 Mb for the two
Formae speciales f.sp.
maclata and f.sp
teres [
121].
5.1. Near-Chromosome-Level Genome Assembly
The chromosome number for fungal species has been observed to range from as few as three (Zolan, 1995) to 21 (haploid) [
117,
122], which is comparable to our reference genome assemblies. The manual observation of our assemblies revealed the presence of TTAGGG repeats that are characteristic of fungal telomeres at the ends of
P. pinodes and
P. pinodella scaffolds. Pulsed-field gel electrophoresis of
A. rabiei isolates from 21 countries, close relatives to the field pea AB species, revealed a chromosome number between 12 and 16. Other techniques such as optical mapping have been used to improve genome assemblies [
123]. Optical mapping can further be used to improve assemblies such as Isolate22Pk with over 200 contigs and a sequencing depth of 32×. However, Isolate3Pp, Isolate18Ppll, Isolate72Ppll, and Isolate87Ppll, with scaffold numbers between 12 and 18 having had deep long-read sequencing coverage up to 190× with correction, even without optical mapping, are near chromosome-level assemblies [
116,
122]. There is a complete lack of descriptions of 1n and 2n forms of the AB fungi in the literature; the original descriptions of the species [
17] and large taxonomic papers [
6] lack this information. Information about ploidy is important in order to carry out genetic engineering and design efficient gene manipulation tools for the AB species. Although AB species ploidy was not estimated in this study, the assemblies generated can be used to determine it using computing tools such as nQuire and the K-mer counting approach [
124,
125]. The number of coding gene sequences (9717–12,249) was also within the range observed for Ascomycota species such as
N. crassa (9820),
A. lentis (11,638), and
L. maculans (12,469). The rRNA and tRNA numbers were comparable to what has been found in the literature for fungal species within the Ascomycota phylum [
116,
117,
126,
127,
128,
129]. Overall, these AB genome assemblies are of high quality, and will be a valuable tool to facilitate the characterization of field pea AB populations, identify evolutionary drivers, and develop diagnostic and monitoring tools that would enable the advancement of durable and targeted management strategies.
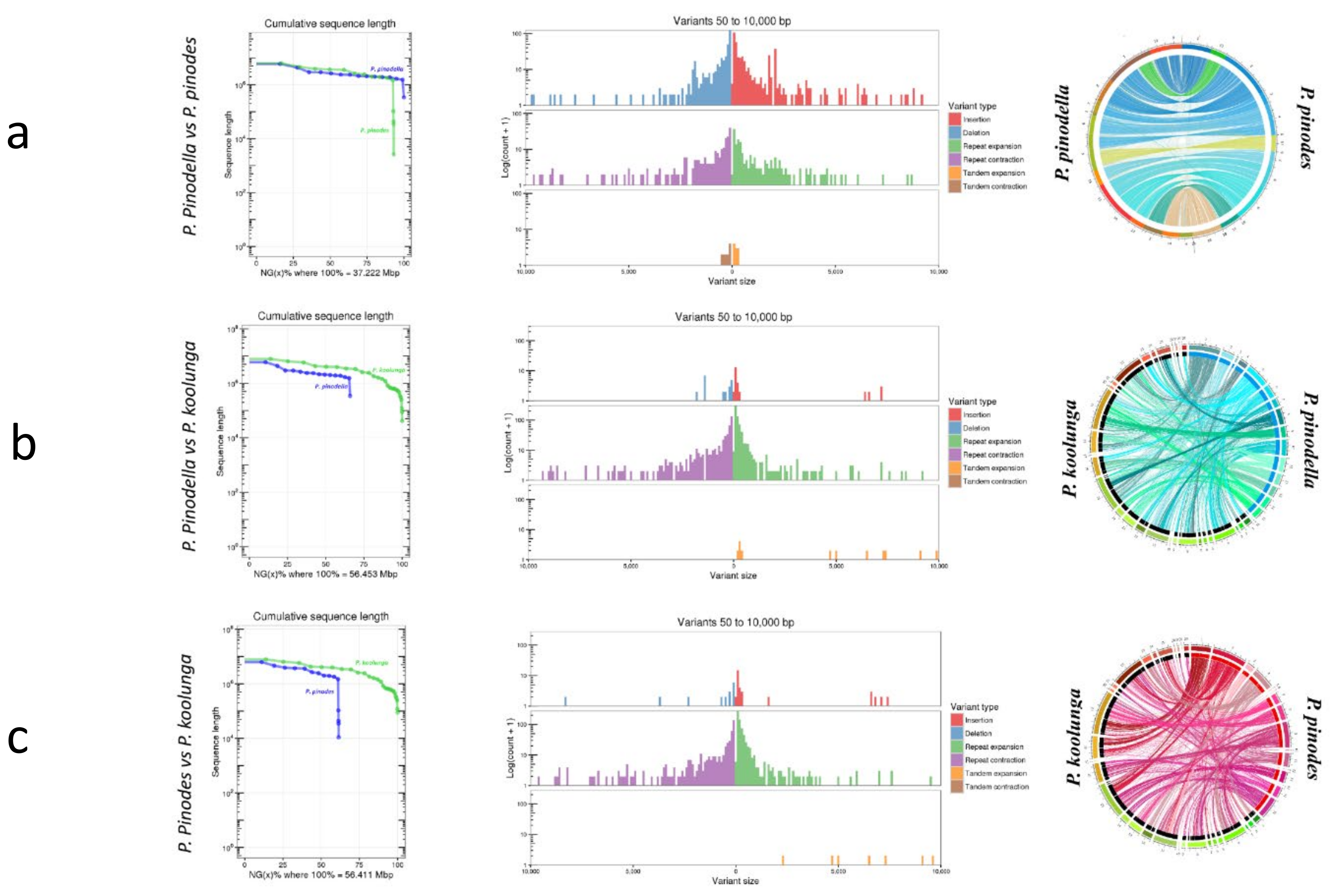
5.2. AB Nuclear Genome Structural Variation
The synteny analysis of our isolates revealed high genomic conservation between species, mostly between P. pinodes and P. pinodella compared to P. koolunga. Host and cultivar specificity in closely related plant pathogenic fungi can be influenced by chromosomal rearrangements and presence–absence variations in key pathogenicity genes.
Chromosomal rearrangements were observed between the AB species, including the sectional translocation of genomic regions among species, which has been observed in other fungi, including
A. rabiei and
A. lentis [
130,
131,
132,
133]. The high repeat expansion observed for
P. koolunga compared with other field pea AB species confirms that the large genome size of
P. koolunga is likely due to large-scale evolution through repeat element expansion, and this could have an impact on pathogenicity and lineage-specificity, as genes near TEs could be altered, giving them new functions [
121,
134]. The presence of species- and isolate-specific TEs among AB species sheds some light on the level of genome diversity possible within AB populations. Simple sequence repeats (SSRs) are the most abundant in the AB genomes, followed by long terminal repeat (LTR) elements such as Gypsy. The most abundant TEs in the literature are the LTR retrotransposons Gypsy and Copia, and DNA Transposons such as Mariner. The percentages of repeat elements and TEs in fungal genomes have been reported to vary significantly among species [
120,
135,
136,
137]. TEs are known to play diverse roles in the genome, including the promotion of genome variability, expansion, increased fitness, and adaptability [
138]. The DNA transposon OPHIO was identified in the
P. pinodella and
P. pinodes genome assemblies. These have been reported to provide genomic responses to biotic and abiotic stressors in the environment, highlighting possible roles in these species [
139]. Further investigation into these TEs will provide clarity on whether they are functional, and information about the specific roles they play in the AB species genome architecture.
The distinct bimodal distribution of GC content bias observed for
P. koolunga and
P. pinodella signifies the presence of large proportions of AT-rich regions, especially in
P. koolunga, which is consistent with the BUSCO GC% obtained for the AB genome assemblies. Highly AT-rich genomes are usually associated with a high content of repeat elements such as transposable elements (TE) [
36,
140]. TEs have been linked to the conferring of advantages in genetic diversity, adaptation and environmental fitness when expressed in the genome [
141]. This gave rise to the “two speed genome” model, which was developed to account for the fact that TEs were observed to be in gene-sparse areas of the genome, and were associated with effector genes [
36,
119,
142]. However, the presence of a high percentage of repeat elements in the genome does not always translate to high expression rates, as we can see in the AB transcriptome repeat analysis results.
P. koolunga, having the highest repeat percentage, seems to have the lowest number of expressed repeats compared to
P. pinodes and
P. pinodella. This could be due to the fungal mechanism designed against the propagation of transposable elements known as repeat-induced point mutation (RIP) [
143,
144]. RIP has been found to deplete GC content, thereby increasing AT content through irreversible transitions of cytosine (C) to thymine (T) [
145]. In silico RIP analysis showed the presence of RIP in all of the field pea AB species analysed, with
P. koolunga having the most extensive RIP (42%) compared to
P. pinodella (11%) and
P. pinodes (3%). This suggests that most of the repeats in the
P. koolunga genome have been silenced, which explains why even though
P. koolunga has the highest repeat content in its assembled genome, it expresses the lowest number of repeat elements in its transcriptome. RIP tends to occur during the sexual phase when dikaryotic cells proliferate [
146]. Extensive RIP is often found in fungi with sexual reproduction capabilities [
147], and given that the sexual stages of
P. pinodella and
P. koolunga have not yet been observed, this result suggests that
P. pinodella and
P. koolunga too might have the capacity for cryptic sexual reproduction, as has been reported for
Aspergillus and
Penicillium species [
146], or might represent ancestral RIP activity, as has been observed in other asexual fungi [
146,
148,
149]. Overall, the differences in the repeat content, both in the genome and transcriptome, suggest the AB pathogens have had different evolutionary trajectories, which should be taken into consideration when developing disease management strategies. The presence of diverse drivers of evolution between field pea AB species would elicit different host response genes among the species upon infection, as has been observed previously [
150].
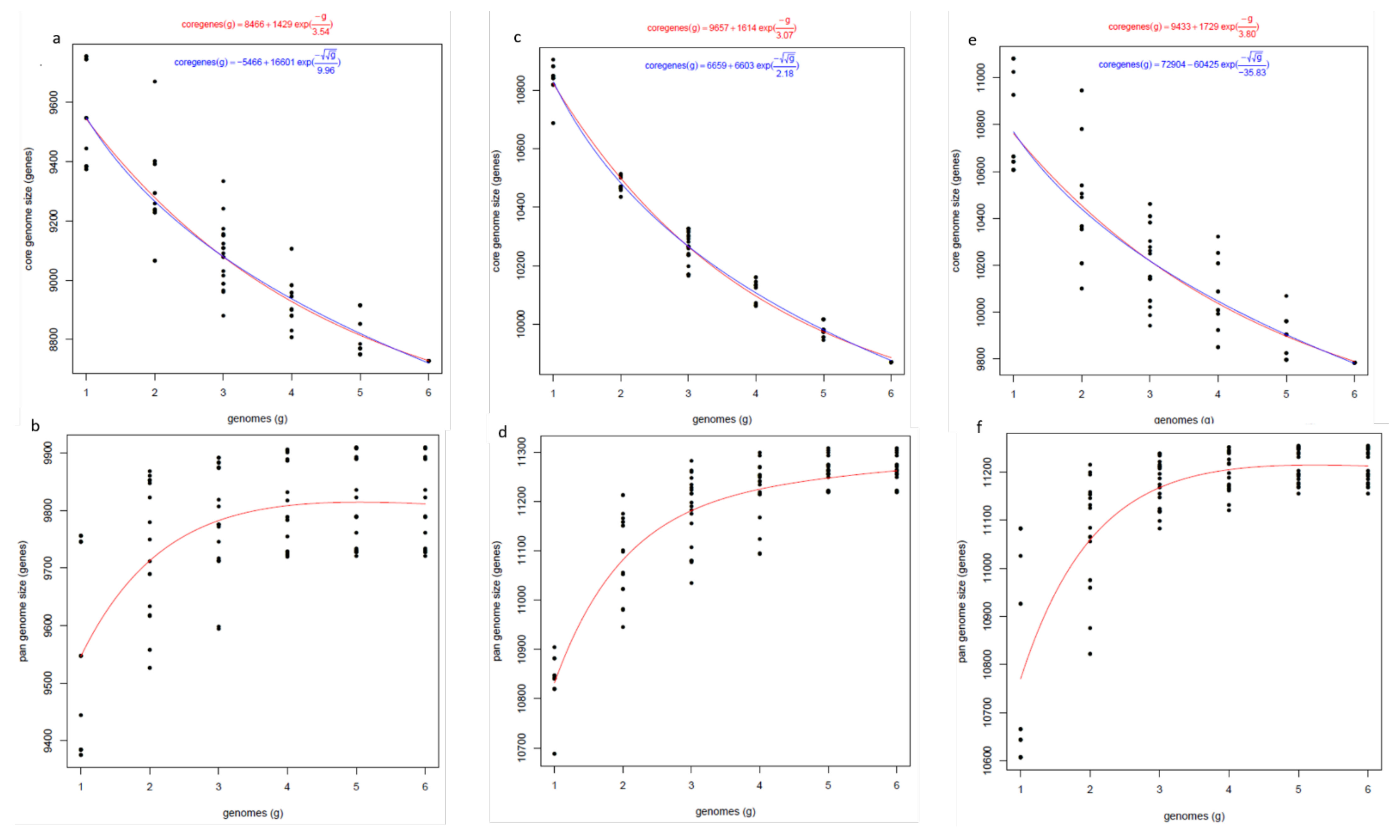
5.3. Orthologous Gene Cluster and Pangenome Analysis
Much more intra-species homology was observed among
P. pinodes isolates compared to
P. pinodella and
P. koolunga.
P. pinodes had more homology with
P. pinodella compared to
P. koolunga, corroborating our results from phylogeny. Pangenome analysis using protein homology did not show significant genome variation among AB species, with about 16% of the genomes accounting for accessory genes. However, some fungal species have been reported to have a higher percentage of accessory genes, such as the 63% reported for
Zymoseptoria tritici [
151]; the 16% observed in the AB species is sufficient to provide diversity in adaptability, virulence and pathogenicity in the field pea AB species, as has been observed for other fungal species, including
Saccharomyces cerevisiae (~14%) and
Aspergillus fumigatus (~16%) [
46]. The gene ontology (GO) analysis of unique orthologous gene clusters revealed genes responsible for various metabolic processes and molecular functions, including pathogenesis. Interestingly, heterokaryon incompatibility protein 6 is unique to
P. pinodes. This protein is responsible for non-self-recognition, which plays a crucial role in the vegetative recognition system in filamentous fungi.
P. pinodes is the only species with a homothallic mating type, and so heterokaryon incompatibility protein 6 could play a role in the regulation of genotypic diversity and gene flow in the species.
5.4. AB Mating Type Determination
Mating type genes have been confirmed previously for
P. pinodella and
P. pinodes [
152,
153,
154].
P. pinodella has been observed to exhibit a heterothallic mating pattern, having either one or the other mating type locus, and
P. pinodes has been observed to be homothallic, having both mating types [
154]. Our results from whole-genome sequencing agree with these findings, with heterothallism being evident for all of the
P. pinodella isolates sequenced, and with homothallism being evident for all of the
P. pinodes isolates. The two idiomorphs of the homothallic
P. pinodes isolates were observed to be on the same scaffold and in close proximity, as has been reported for homothallic ascomycete species in
Gibberella zeae [
155] and
Chaetomium globosum [
156]. On the other hand, the mating type for
P. koolunga has not been defined previously, and from our analysis we can conclude that the species exhibits a heterothallic mating pattern. This is similar to the mating type patterns observed for
A. rabiei,
A. lentis and
A. fabae, which show a very close phylogenetic relationship to
P. koolunga, as well as Phoma species such as
Phoma clematidina [
117,
154,
157,
158,
159].
5.5. Phylogeny
The congruence observed between the whole-genome and ortholog inference phylogeny of AB species showed a clear separation of the species, particularly P. pinodella and P. pinodes. The clear speciation of the AB species inferred in the phylogenetic analysis implies there is enough genetic variation between these species to be able to develop molecular markers for diagnostics. Previous research has reported the inability to differentiate P. pinodella and P. pinodes using ITS, and our results confirm this, showing P. pinodella and P. pinodes clustering together in the ITS dendrogram. Although protein coding genes have been proven to give a better resolution of the phylogeny, not all of them are able to provide a high resolution for all of the fungal species. Our results showed the low resolution of the species observed for RPB2 and Beta tubulin. These AB species were classified using a CAPS assay based on RPB2 gene, and the misidentification of some of the isolates has been observed, signifying the low resolution of the RPB2 gene for AB species P. pinodella and P. pinodes. Tef1 and TOP1 displayed the high species resolution of P. pinodella and P. pinodes, making them strong candidates as barcoding markers for AB species identification.
Gene duplication events have been known to play important roles in the evolution of species by giving rise to new genes that code for proteins with potentially novel functions, which could include increased fitness and phenotypic diversity [
160,
161,
162,
163]. Here, we see that gene duplication played a major role in the speciation of the AB species separating
P. pinodes and
P. pinodella. Further analysis can be carried out to identify the specific genes duplicated, the resulting genomic structure, and the impact of the duplication events.
5.6. CAZyme Analysis
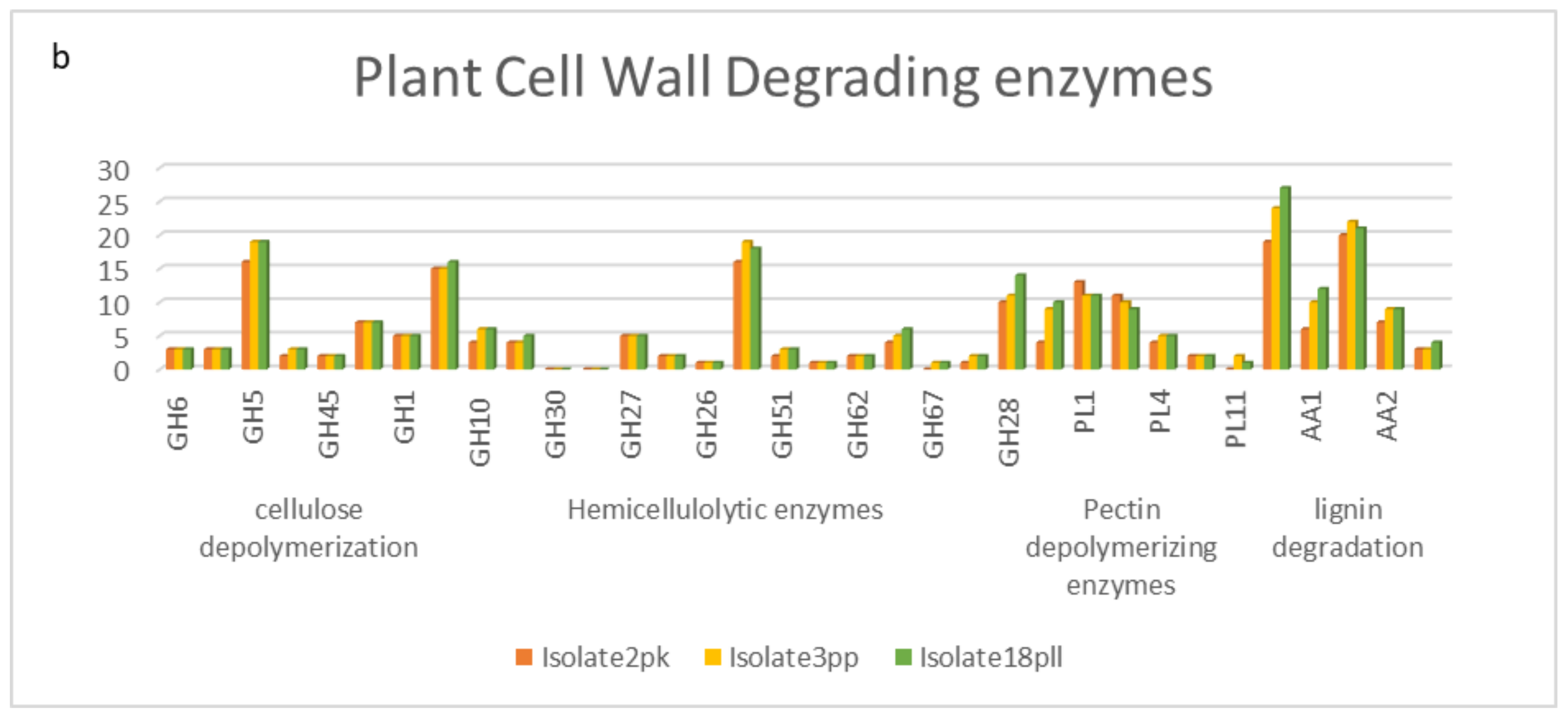
Predicted CAZymes also fall within the range of number and class distributions reported for plant necrotrophic fungi, with approximately 400–850 CAZymes in total, which is in line with the observed necrotrophic lifestyle for the field pea AB isolates [
164]. The analysis of specific cell wall degrading enzymes reveals a high number of cellulose depolymerization, hemicellulolytic, pectin depolymerization and lignin degradation enzymes signifying major food sources for AB fungus. Pea has been reported to contain 32% hemicellulose, 27% cellulose, 41% pectin and at least 5.3% lignin. The AB species cell wall degradation repertoire reflects the enzymes that are responsible for successful pathogenicity of the pea plant [
165,
166]. An increased host range is equally proportional to the diversity of CAZymes in the fungal genome, as has been described for
Macrophomina phaseolina (Islam MS, et al., 2012). The large CAZyme repertoire for the AB species suggests a broad host range, which is in congruence with host specificity experiments carried out for the species [
167,
168,
169,
170].
5.7. Secondary Metabolite Analysis
Fungal secondary metabolites (SMs) have been reported to play significant roles in pathogenicity, infection, and host specificity in necrotrophs [
171,
172,
173]. Biosynthesis of fungal SMs is carried out by co-regulated genes, usually clustered at a genomic locus referred to as a biosynthetic gene cluster (BGCs). AB species’ genome-wide BCGs are in the range observed for
C. fulvum (23) and
S. sclerotiorum (29) which are also plant pathogenic fungi [
174,
175]. Ascomycete-specific PKS-NRPs hybrids were present in all of the AB isolates, and this agrees with their phylum identification [
176]. Hybrid metabolites are known to be common in fungi, and we observed a few of them with moieties from different classes, including NRPS, T1PKS, and indole observed in one
P. koolunga isolate.
Fungal-RiPPs are a class of BGCs that produce metabolites with a diverse variety of bioactivities involved in defence, competition, communication, and virulence [
177,
178,
179,
180]. RiPPs have been extensively characterized in plants and bacteria but are underexplored in fungi, although their first identification was in a fungus [
181]. Fungal-RiPPs were observed in
P. pinodella and
P. pinodes species, indicating a possible role specific to these species compared to
P. koolunga. Further characterization would be needed in order to identify the biosynthetic potential of these BCGs in these species. β-Lactones are also an underexplored class of fungal BGCs linked to several antimicrobial activities [
182], and were also found to be specific to
P. pinodella and
P. pinodes species.
Terpenes and terpenoids play diverse roles in fungi: they can function as toxins, promote growth, and have antifungal properties [
176,
183]. The high production of terpenes has been observed in
Phoma sp., which along with other volatile organic chemicals exhibit antifungal properties to certain fungal species, which is a possible defence mechanism [
184]. However, it is unsurprising that
P. koolunga, also a
Phoma sp., exhibited a higher production of terpenes compared to the other AB species. Consequently, all of the AB species can be present on the plant at the same time, and so our results imply that the SMs repertoire of the individual AB species could either be tolerant to the other AB species in the complex or could serve to reduce competition; the latter has been reported [
185].
Overall, the SMs diversity observed for the field pea AB species reveals that
P. pinodella and
P. pinodes species are more alike in their SM repertoire compared to
P. koolunga. Although their host specificity overlaps, they seem to deploy different bioactive mechanisms, which could be why they elicit different host response genes, and resistance to one species does not translate to resistance to other species within the complex.
P. pinodes is a broad host range fungus, being able to infect numerous legumes [
169]; however, no research on host range specificity has been conducted for
P. pinodella. Given the high genomic similarity between these two AB species,
P. pinodella could possibly have a similar host range beyond field pea, similarly to
P. pinodes.
5.8. Mitochondrial Genome Assembly
Although the mitochondrial genome size varied among the species and isolates, the numbers fall within the reported mitochondrial genomes for Ascomycota species [
116,
117,
186,
187,
188,
189]. There is a positive correlation between the mitochondrial genome length and number of introns. The genome size for
P. koolunga was close to that reported for closely related species
A. lentis and
A. rabiei. The gene order observed shows high synteny within AB species. The number of protein coding genes observed is within that reported for several fungi [
190,
191]. Recent research characterizing introns and their HEGs across the fungi phyla reveal varying numbers of introns in protein coding genes, with the highest number in the
cox1 gene, followed by
cob and
nad5 [
190]. Genes with introns within AB species include
cox1,
cox2,
cox3,
cob,
nad1,
nad5 and
nad4L. Similar gene interruptions have been reported for
Aspergillus sp.,
Saccharomyces sp.,
Podospora sp.,
Neurospora sp. and
Penicillium sp. [
188,
190,
192,
193,
194]. Interestingly, potential trans-splicing mediated by group 1 introns was observed within
cox1,
cox2,
nad5,
cob and
nad2 genes across all of the AB species. Trans-splicing has been reported in animals, plants, green algae, and fungi [
195,
196,
197,
198,
199,
200,
201]. Most of these reports are for
cox1 genes, and nothing has been reported for
cox2,
nad5,
cob and
nad2, although Cis-spliced group 1 introns have been observed in
cox1 and
nad5 in fungi [
202]. Additional analysis would be required in order to confirm this based on evidence of a ligated mature mRNA at exon–intron junctions and a complete intron RNA structure [
195]. Furthermore, the presence of T7 phage-like single-subunit RNA polymerase (
rpo) in only
P. koolunga species suggests the insertion of a plasmid into its mitochondrial DNA. These
rpo genes have been identified in other ascomycete fungi, and they are reported to be most likely non-functional, possibly fragmented, and eventually lost (Lang 2018). Previous research found that rpo was only found in species belonging to the fungal phyla Ascomycota, Basidiomycota and Chytridiomycota. Although rpo homologs were found in both
Ascomycota and
Basidiomycota fungal species, this was only in 10% of the Ascomycota species (227) sampled, compared to 57% for
Basidiomycota (116) [
203]. This means that
P. koolunga is among a small percentage of species within the Ascomycota phylum that harbours an rpo homolog. Rpo-based phylogeny revealed both phyla-specific clades and rpo sequences that are shared between species of different phyla. This diversity of rpo sequences observed in the literature suggests a possible horizontal gene transfer between fungal species [
203,
204,
205]. The mitochondrial codon usage for the AB species is similar to that reported for other ascomycete fungi [
187]. A preference for AT-rich codons contributes to the low GC content (28–29%) in the AB species which has been observed for
Aspergillus sp. and
Penicillium sp. [
188].
Phylogenetic analysis of the AB mitochondrial genomes reveals clear species separation, which can be further explored as barcoding markers for the species. This can be a standalone marker depending on its specificity to the species, or in addition to the other protein coding gene markers highlighted.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}