
Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges



Abstract
:1. Introduction
2. Materials and Methods
2.1. Databases
2.1.1. Details and Rationales of Experimental Structural Templates Used for Docking and Dynamics
2.1.2. Dataset of Coronavirus Spikes
2.2. Modeling of Missing Disordered Loops in Spike
2.3. The Spike–Furin Molecular Docking Simulations
2.3.1. Blind ab Initio Docking in Cluspro 2.0
2.3.2. Cross-Validation by Guided Docking in ‘Zdock + IRaPPA Re-Ranking’
2.3.3. Setting Up Appropriate Baselines: The SARS-CoV Spike–Furin Guided Docking in ‘Zdock + IRaPPA Re-Ranking’
2.3.4. Docking Scoring, Ranking and Re-Ranking
2.3.4.1. Buried Surface Area Calculations
2.3.4.2. Shape Complementarity
2.3.4.3. The Sdock Score
2.4. Molecular Dynamic Simulations
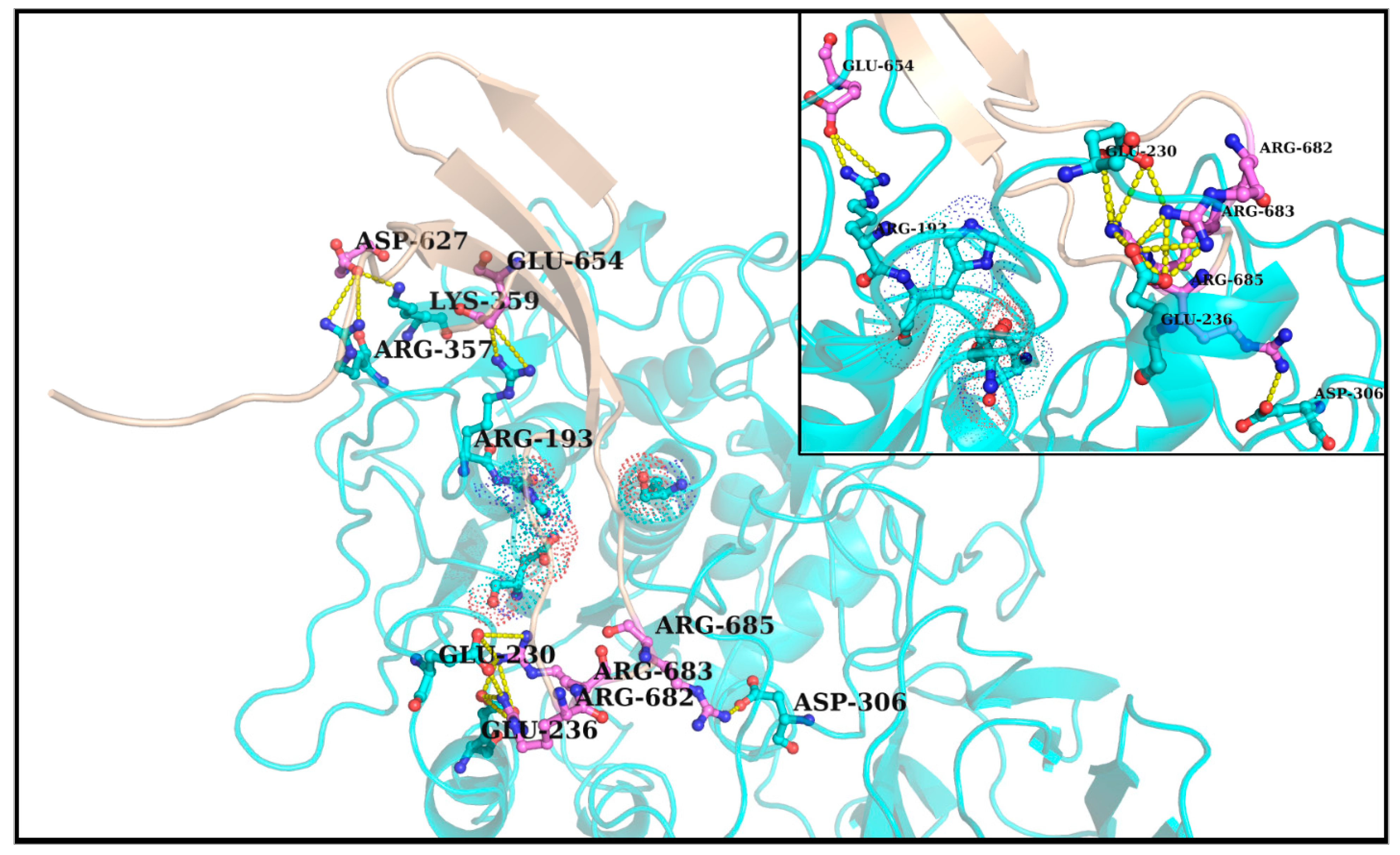
2.5. Identifying Salt-Bridges at the Spike–Furin Interface
2.5.1. Analyzing Salt-Bridge Dynamics
2.5.1.1. Salt-Bridge Persistence and Occurrence
2.5.1.2. Average Contact Intensities of Salt-Bridges
2.6. Calculation of Structure-Based Equilibrium Thermodynamic Parameters (∆H, ∆S, ∆G) for the Spike–Furin Binding
2.7. Ramachandran Plot (RP) Derived Parameters to Probe State-Transitions (e.g., Disorder-to-Order)
2.8. Quantifying a Change between Two N-Binned Frequency Distributions and Assessing Its Statistical Significance in Terms of χ2
3. Results and Discussion
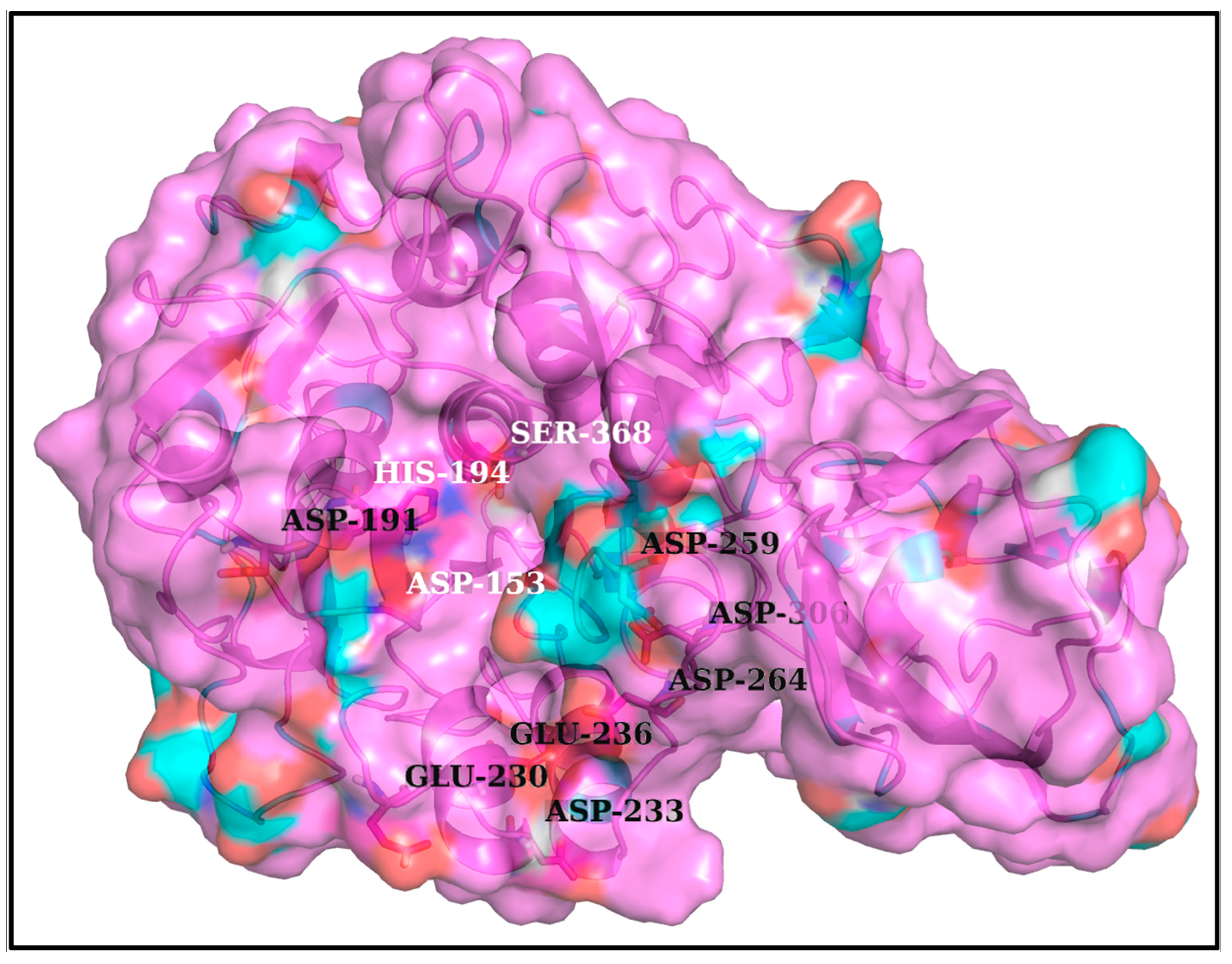
3.1. Structural Insight into the Furin Cleavage Mechanisms
3.2. More the Arginines, More the Disorder’ in the FLCSSpike Activation Loops
3.3. Filling Up the Voids in the Spike Structures: The FLCSSpike Disordered Ensembles
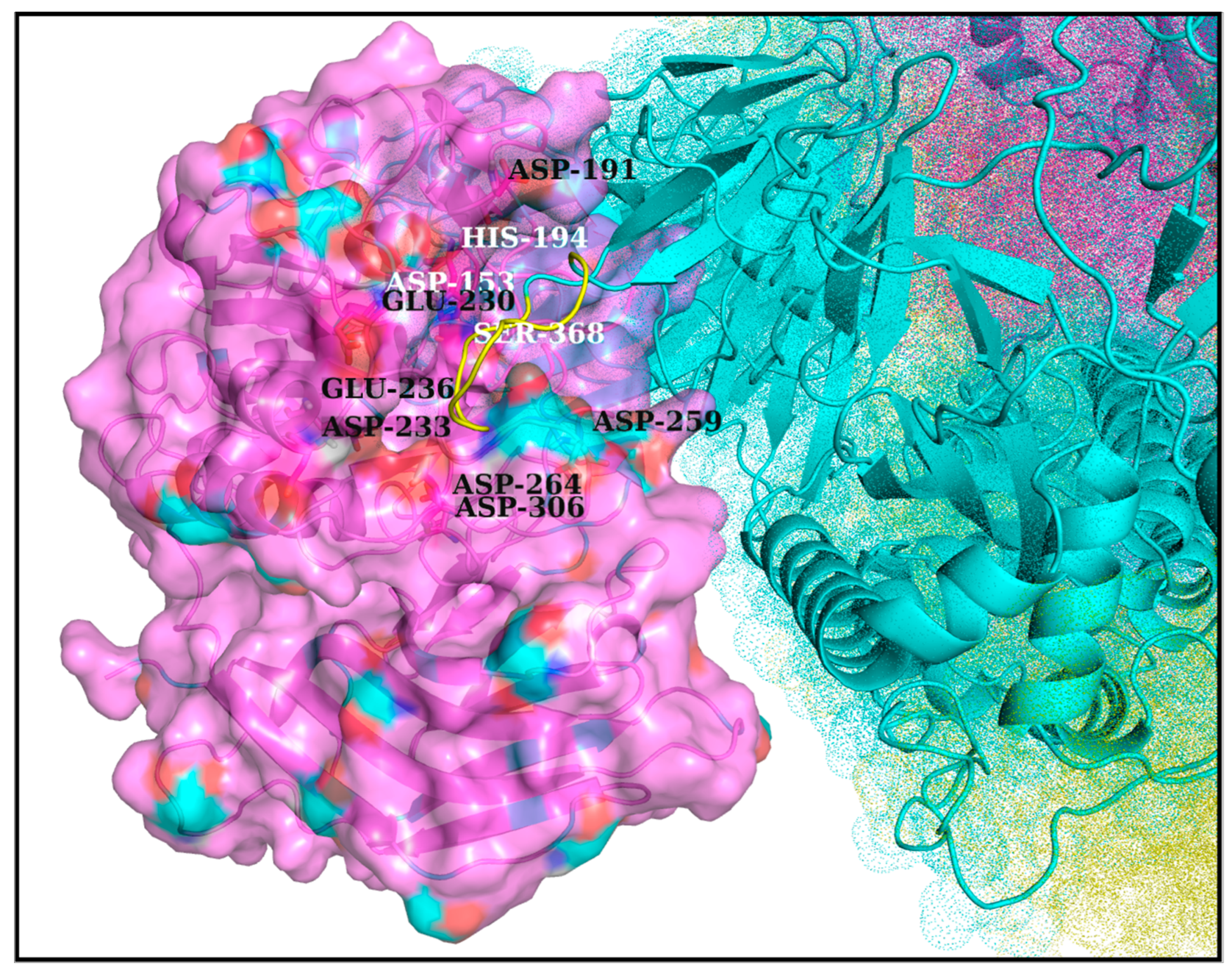
3.4. Docking Furin onto Spike: Using the Pentapeptide Activation Loop to Filter and Accumulate Correctly Docked Poses
3.5. Plausible ‘Disorder-to-Order’ Transition Triggered by Salt-Bridge Dynamics at the Spike–Furin Interface: The ‘Salt-Bridge Hypothesis’
3.6. Validations and Cross-Validations of the ‘Salt-Bridge Hypothesis’
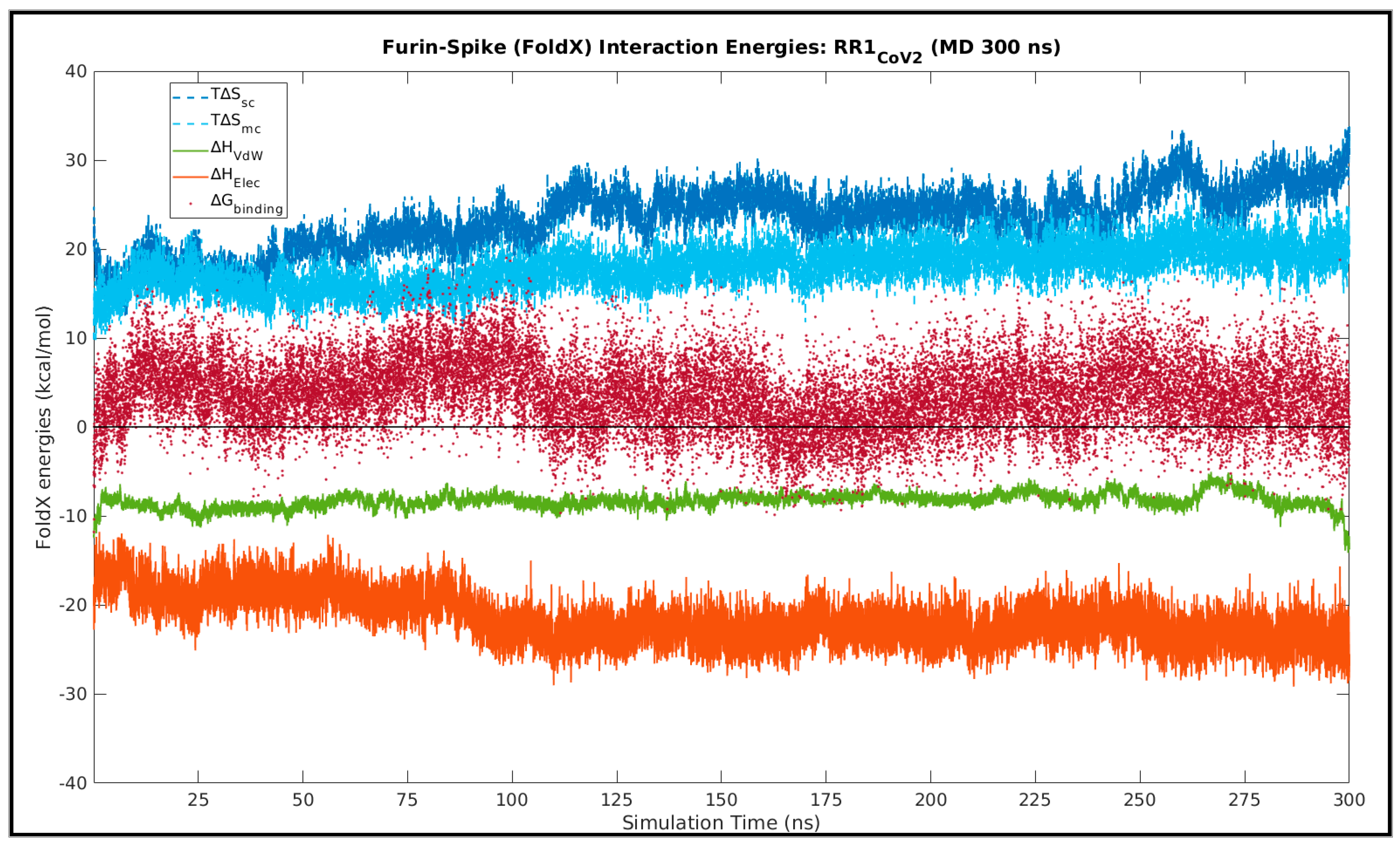
3.6.1. In RR1CoV-2
3.6.2. In ZR1CoV-2
3.6.3. In ZR1CoV, the Baseline
3.7. Enthalpy Entropy Compensation Involved in the Spike–Furin Interaction
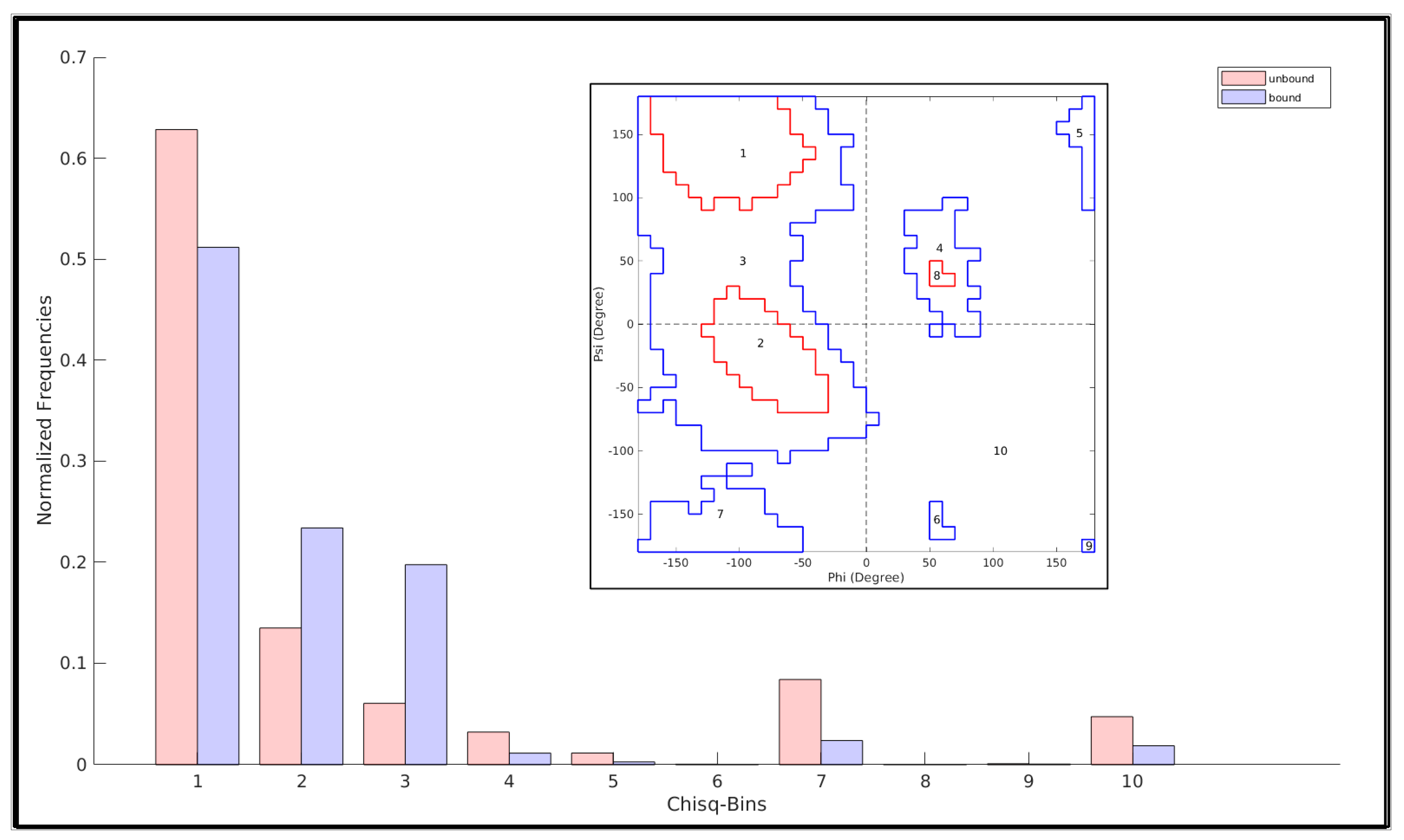
3.8. Using the Ramachandran Plot to Probe the ‘Disorder-to-Order Transition’ of the SARS-CoV-2 FLCSSpike Loop upon Furin Binding
4. Conclusions and Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balaram, P. The Murky Origins of the Coronavirus SARS-CoV-2, the Causative Agent of the COVID-19 Pandemic. Curr. Sci. 2021, 120, 4. [Google Scholar]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Zhan, S.H. The Emergence of the Spike Furin Cleavage Site in SARS-CoV-2. Mol. Biol. Evol. 2021, 39, msab327. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calisher, C.; Carroll, D.; Colwell, R.; Corley, R.B.; Daszak, P.; Drosten, C.; Enjuanes, L.; Farrar, J.; Field, H.; Golding, J.; et al. Statement in Support of the Scientists, Public Health Professionals, and Medical Professionals of China Combatting COVID-19. Lancet 2020, 395, e42–e43. [Google Scholar] [CrossRef] [Green Version]
- Wade, N. The Origin of COVID: Did People or Nature Open Pandora’s Box at Wuhan? 2021. Available online: https://thebulletin.org/2021/05/the-origin-of-covid-did-people-or-nature-open-pandoras-box-at-wuhan/ (accessed on 29 December 2021).
- Malaiyan, J.; Arumugam, S.; Mohan, K.; Gomathi Radhakrishnan, G. An Update on the Origin of SARS-CoV-2: Despite Closest Identity, Bat (RaTG13) and Pangolin Derived Coronaviruses Varied in the Critical Binding Site and O-Linked Glycan Residues. J. Med. Virol. 2021, 93, 499–505. [Google Scholar] [CrossRef]
- Casadevall, A.; Weiss, S.R.; Imperiale, M.J. Can Science Help Resolve the Controversy on the Origins of the SARS-CoV-2 Pandemic? mBio 2021, 12, e01948-21. [Google Scholar] [CrossRef]
- Frutos, R.; Gavotte, L.; Devaux, C.A. Understanding the Origin of COVID-19 Requires to Change the Paradigm on Zoonotic Emergence from the Spillover to the Circulation Model. Infect. Genet. Evol. 2021, 95, 104812. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Imperiale, M.J.; Casadevall, A. Rethinking Gain-of-Function Experiments in the Context of the COVID-19 Pandemic. mBio 2020, 11, e01868-20. [Google Scholar] [CrossRef]
- Bloom, J.D.; Chan, Y.A.; Baric, R.S.; Bjorkman, P.J.; Cobey, S.; Deverman, B.E.; Fisman, D.N.; Gupta, R.; Iwasaki, A.; Lipsitch, M.; et al. Investigate the Origins of COVID-19. Science 2021, 372, 694. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Jiang, J.-Z.; Wan, X.-F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are Pangolins the Intermediate Host of the 2019 Novel Coronavirus (SARS-CoV-2)? PLoS Pathog. 2020, 16, e1008421. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Q.; Yuan, X.; Li, X.; Zhu, T.; Ma, Y.; Chen, J.-L. Analysis of the Codon Usage Pattern in Middle East Respiratory Syndrome Coronavirus. Oncotarget 2017, 8, 110337–110349. [Google Scholar] [CrossRef] [Green Version]
- Das, J.K.; Roy, S. Comparative Analysis of Human Coronaviruses Focusing on Nucleotide Variability and Synonymous Codon Usage Patterns. Genomics 2021, 113, 2177–2188. [Google Scholar] [CrossRef]
- Basu, S.; Chakravarty, D.; Bhattacharyya, D.; Saha, P.; Patra, H.K. Plausible Blockers of Spike RBD in SARS-CoV2—Molecular Design and Underlying Interaction Dynamics from High-Level Structural Descriptors. J. Mol. Model. 2021, 27, 191. [Google Scholar] [CrossRef]
- Chowdhury, R.; Boorla, V.S.; Maranas, C.D. Computational Biophysical Characterization of the SARS-CoV-2 Spike Protein Binding with the ACE2 Receptor and Implications for Infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 2573–2582. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins. Act. Viruses Host Proteases 2018, 16, 71–98. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Whittaker, G.R. Physiological and Molecular Triggers for SARS-CoV Membrane Fusion and Entry into Host Cells. Virology 2018, 517, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM Structures of MERS-CoV and SARS-CoV Spike Glycoproteins Reveal the Dynamic Receptor Binding Domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-Electron Microscopy Structures of the SARS-CoV Spike Glycoprotein Reveal a Prerequisite Conformational State for Receptor Binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xiang, R.; Huo, S.; Zhou, Y.; Jiang, S.; Wang, Q.; Yu, F. Molecular Mechanism of Interaction between SARS-CoV-2 and Host Cells and Interventional Therapy. Signal Transduct. Target. Ther. 2021, 6, 1–19. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin Cleavage of SARS-CoV-2 Spike Promotes but Is Not Essential for Infection and Cell-Cell Fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; Consortium, P.V.; de Silva, T.I.; Barclay, W.S. The SARS-CoV-2 Variants Associated with Infections in India, B.1.617, Show Enhanced Spike Cleavage by Furin. BioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.05.28.446163v1.abstract (accessed on 29 December 2021).
- Nagy, A.; Basiouni, S.; Parvin, R.; Hafez, H.M.; Shehata, A.A. Evolutionary Insights into the Furin Cleavage Sites of SARS-CoV-2 Variants from Humans and Animals. Arch. Virol. 2021, 166, 2541–2549. [Google Scholar] [CrossRef]
- How Ominous Is the Omicron Variant (B.1.1.529)? Available online: https://asm.org/Articles/2021/December/How-Ominous-is-the-Omicron-Variant-B-1-1-529 (accessed on 27 December 2021).
- Bertram, S.; Glowacka, I.; Müller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by Human Airway Trypsin-like Protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, S.; Cameron, A.; Bourenkov, G.P.; Kiefersauer, R.; Huber, R.; Lindberg, I.; Bode, W.; Than, M.E. The Crystal Structure of the Proprotein Processing Proteinase Furin Explains Its Stringent Specificity. Nat. Struct. Biol. 2003, 10, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of Furin Cleavage Site Attenuates SARS-CoV-2 Pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Mustafa, Z.; Zhanapiya, A.; Kalbacher, H.; Burster, T. Neutrophil Elastase and Proteinase 3 Cleavage Sites Are Adjacent to the Polybasic Sequence within the Proteolytic Sensitive Activation Loop of the SARS-CoV-2 Spike Protein. ACS Omega 2021, 6, 7181–7185. [Google Scholar] [CrossRef]
- Chaudhry, M.Z.; Eschke, K.; Hoffmann, M.; Grashoff, M.; Abassi, L.; Kim, Y.; Brunotte, L.; Ludwig, S.; Kröger, A.; Klawonn, F.; et al. Rapid SARS-CoV-2 Adaptation to Available Cellular Proteases. J. Virol. 2022, 2022, jvi0218621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Wu, J.; Yu, Y.; Liu, S.; Li, T.; Li, Q.; Ding, R.; Wang, H.; Nie, J.; et al. A Second Functional Furin Site in the SARS-CoV-2 Spike Protein. Emerg. Microbes Infect. 2022, 11, 182–194. [Google Scholar] [CrossRef]
- Lemmin, T.; Kalbermatter, D.; Harder, D.; Plattet, P.; Fotiadis, D. Structures and Dynamics of the Novel S1/S2 Protease Cleavage Site Loop of the SARS-CoV-2 Spike Glycoprotein. J. Struct. Biol. X 2020, 4, 100038. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmer, J.C.; Zhu, W.; Pop, C.; Regan, T.; Snipas, S.J.; Eroshkin, A.M.; Riedl, S.J.; Salvesen, G.S. Structural and Kinetic Determinants of Protease Substrates. Nat. Struct. Mol. Biol. 2009, 16, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E.; Alves, J.; Garay-Malpartida, M.; Occhiucci, J.M. Coupling Caspase Cleavage and Proteasomal Degradation of Proteins Carrying PEST Motif. Curr. Protein Pept. Sci. 2008, 9, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Örd, M.; Faustova, I.; Loog, M. The Sequence at Spike S1/S2 Site Enables Cleavage by Furin and Phospho-Regulation in SARS-CoV2 but Not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 16944. [Google Scholar] [CrossRef]
- Gagliardi, P.A.; Primo, L. Irreversible Activation of Rho-Activated Kinases Resulted from Evolution of Proteolytic Sites within Disordered Regions in Coiled-Coil Domain. Mol. Biol. Evol. 2019, 36, 376–392. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC14: International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Huang, P.-S.; Ban, Y.-E.A.; Richter, F.; Andre, I.; Vernon, R.; Schief, W.R.; Baker, D. RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design. PLoS ONE 2011, 6, e24109. [Google Scholar] [CrossRef] [Green Version]
- Mandell, D.J.; Coutsias, E.A.; Kortemme, T. Sub-Angstrom Accuracy in Protein Loop Reconstruction by Robotics-Inspired Conformational Sampling. Nat. Methods 2009, 6, 551–552. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Söderquist, F.; Wallner, B. Proteus: A Random Forest Classifier to Predict Disorder-to-Order Transitioning Binding Regions in Intrinsically Disordered Proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef]
- Vankadari, N. Structure of Furin Protease Binding to SARS-CoV-2 Spike Glycoprotein and Implications for Potential Targets and Virulence. J. Phys. Chem. Lett. 2020, 11, 6655–6663. [Google Scholar] [CrossRef]
- Basu, S.; Biswas, P. Salt-Bridge Dynamics in Intrinsically Disordered Proteins: A Trade-off between Electrostatic Interactions and Structural Flexibility. Biochim. Biophys. Acta BBA-Proteins Proteom. 2018, 1866, 624–641. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Basu, S. Criticality in the Conformational Phase Transition among Self-Similar Groups in Intrinsically Disordered Proteins: Probed by Salt-Bridge Dynamics. Biochim. Biophys. Acta BBA-Proteins Proteom. 2020, 1868, 140474. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of Polypeptide Chain Configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor Binding and Priming of the Spike Protein of SARS-CoV-2 for Membrane Fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Seidah, N.G.; Mayer, G.; Zaid, A.; Rousselet, E.; Nassoury, N.; Poirier, S.; Essalmani, R.; Prat, A. The Activation and Physiological Functions of the Proprotein Convertases. Int. J. Biochem. Cell Biol. 2008, 40, 1111–1125. [Google Scholar] [CrossRef]
- Dahms, S.O.; Arciniega, M.; Steinmetzer, T.; Huber, R.; Than, M.E. Structure of the Unliganded Form of the Proprotein Convertase Furin Suggests Activation by a Substrate-Induced Mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, 11196–11201. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of Proprotein Convertase Cleavage Sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Fedry, J.; Hurdiss, D.L.; Wang, C.; Li, W.; Obal, G.; Drulyte, I.; Du, W.; Howes, S.C.; van Kuppeveld, F.J.M.; Förster, F.; et al. Structural Insights into the Cross-Neutralization of SARS-CoV and SARS-CoV-2 by the Human Monoclonal Antibody 47D11. Sci. Adv. 2021, 7, eabf5632. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L. The Convalescent Sera Option for Containing COVID-19. J. Clin. Investig. 2020, 130, 1545–1548. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM Structure of the SARS Coronavirus Spike Glycoprotein in Complex with Its Host Cell Receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural Impact on SARS-CoV-2 Spike Protein by D614G Substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5.6.1–5.6.30. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-Based Protein Docking Program with Pairwise Potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez, R.; Leplae, R.; De Maria, L.; Wodak, S.J. Assessment of Blind Predictions of Protein–Protein Interactions: Current Status of Docking Methods. Proteins Struct. Funct. Bioinform. 2003, 52, 51–67. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers. Bioinform. Oxf. Engl. 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Hubbard, S.; Thornton, J.; NACCESS. Computer Program, Department of Biochemistry and Molecular Biology, University College London—Open Access Library. 1993. Available online: http://www.oalib.com/references/5299711 (accessed on 1 March 2017).
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Basu, S.; Wallner, B. Finding Correct Protein-Protein Docking Models Using ProQDock. Bioinformatics 2016, 32, i262–i270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Sen, M.; Bhattacharya, D.; Saha, P. The Jigsaw Puzzle Model: Search for Conformational Specificity in Protein Interiors. J. Mol. Biol. 2003, 333, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Mapping the Distribution of Packing Topologies within Protein Interiors Shows Predominant Preference for Specific Packing Motifs. BMC Bioinform. 2011, 12, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Self-Complementarity within Proteins: Bridging the Gap between Binding and Folding. Biophys. J. 2012, 102, 2605–2614. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Applications of Complementarity Plot in Error Detection and Structure Validation of Proteins. Indian J. Biochem. Biophys. 2014, 51, 188–200. [Google Scholar]
- Lawrence, M.C.; Colman, P.M. Shape Complementarity at Protein/Protein Interfaces. J. Mol. Biol. 1993, 234, 946–950. [Google Scholar] [CrossRef]
- Basu, S. CPdock: The Complementarity Plot for Docking of Proteins: Implementing Multi-Dielectric Continuum Electrostatics. J. Mol. Model. 2017, 24, 8. [Google Scholar] [CrossRef]
- SC (CCP4: Supported Program)—CCP4Docs Documentation. Available online: https://www.ccp4.ac.uk/html/sc.html (accessed on 11 November 2021).
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Siu, S.W.I.; Pluhackova, K.; Böckmann, R.A. Optimization of the OPLS-AA Force Field for Long Hydrocarbons. J. Chem. Theory Comput. 2012, 8, 1459–1470. [Google Scholar] [CrossRef]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield: Implications for Immune Recognition. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Basu, S.; Mukharjee, D. Salt-Bridge Networks within Globular and Disordered Proteins: Characterizing Trends for Designable Interactions. J. Mol. Model. 2017, 23, 206. [Google Scholar] [CrossRef] [PubMed]
- Musafia, B.; Buchner, V.; Arad, D. Complex Salt Bridges in Proteins: Statistical Analysis of Structure and Function. J. Mol. Biol. 1995, 254, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M.; Gally, J.A. Degeneracy and Complexity in Biological Systems. Proc. Natl. Acad. Sci. USA 2001, 98, 13763–13768. [Google Scholar] [CrossRef] [Green Version]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Simonetti, F.L.; Goncearenco, A.; Panchenko, A.R. MutaBind Estimates and Interprets the Effects of Sequence Variants on Protein–Protein Interactions. Nucleic Acids Res. 2016, 44, W494–W501. [Google Scholar] [CrossRef] [Green Version]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Broom, A.; Jacobi, Z.; Trainor, K.; Meiering, E.M. Computational Tools Help Improve Protein Stability but with a Solubility Tradeoff. J. Biol. Chem. 2017, 292, 14349–14361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhee, P.; Verschueren, E.; Baeten, L.; Stricher, F.; Serrano, L.; Rousseau, F.; Schymkowitz, J. BriX: A Database of Protein Building Blocks for Structural Analysis, Modeling and Design. Nucleic Acids Res. 2011, 39, D435–D442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamisetty, H.; Ramanathan, A.; Bailey-Kellogg, C.; Langmead, C.J. Accounting for Conformational Entropy in Predicting Binding Free Energies of Protein-Protein Interactions. Proteins 2011, 79, 444–462. [Google Scholar] [CrossRef]
- Brackley, C.A.; Liebchen, B.; Michieletto, D.; Mouvet, F.; Cook, P.R.; Marenduzzo, D. Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains. Biophys. J. 2017, 112, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, J.-M.; Gibrat, G.; Bellissent-Funel, M.-C. Hydration Water Rotational Motion as a Source of Configurational Entropy Driving Protein Dynamics. Crossovers at 150 and 220 K. Phys. Chem. Chem. Phys. 2008, 10, 4865–4870. [Google Scholar] [CrossRef]
- Angell, C.A. Entropy and Fragility in Supercooling Liquids. J. Res. Natl. Inst. Stand. Technol. 1997, 102, 171–185. [Google Scholar] [CrossRef]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a PH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.e5. [Google Scholar] [CrossRef]
- Cueno, M.E.; Ueno, M.; Iguchi, R.; Harada, T.; Miki, Y.; Yasumaru, K.; Kiso, N.; Wada, K.; Baba, K.; Imai, K. Insights on the Structural Variations of the Furin-Like Cleavage Site Found Among the December 2019–July 2020 SARS-CoV-2 Spike Glycoprotein: A Computational Study Linking Viral Evolution and Infection. Front. Med. 2021, 8, 240. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise Disordered Region Predictions with Annotated Protein-Binding Activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of Disordered Protein Regions from Amino Acid Sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Katuwawala, A.; Kurgan, L. Comparative Assessment of Intrinsic Disorder Predictions with a Focus on Protein and Nucleic Acid-Binding Proteins. Biomolecules 2020, 10, 1636. [Google Scholar] [CrossRef] [PubMed]
- Katuwawala, A.; Oldfield, C.J.; Kurgan, L. Accuracy of Protein-Level Disorder Predictions. Brief Bioinform. 2020, 21, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.T.C. Electrostatics in Intrinsically Disordered Proteins. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2012. [Google Scholar]
- Liu, C.; Wang, T.; Bai, Y.; Wang, J. Electrostatic Forces Govern the Binding Mechanism of Intrinsically Disordered Histone Chaperones. PLoS ONE 2017, 12, e0178405. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Bahadur, R.P. Conservation and Coevolution Determine Evolvability of Different Classes of Disordered Residues in Human Intrinsically Disordered Proteins. Proteins Struct. Funct. Bioinform. 2021, 90, 632–644. [Google Scholar] [CrossRef]
- Wong, E.T.C.; Na, D.; Gsponer, J. On the Importance of Polar Interactions for Complexes Containing Intrinsically Disordered Proteins. PLoS Comput. Biol. 2013, 9, e1003192. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, G.; Salladini, E.; Santambrogio, C.; Grandori, R.; Longhi, S.; Brocca, S. Conformational Response to Charge Clustering in Synthetic Intrinsically Disordered Proteins. Biochim. Biophys. Acta BBA-Gen. Subj. 2018, 1862, 2204–2214. [Google Scholar] [CrossRef]
- Cui, H.; Eltoukhy, L.; Zhang, L.; Markel, U.; Jaeger, K.-E.; Davari, M.D.; Schwaneberg, U. Less Unfavorable Salt Bridges on the Enzyme Surface Result in More Organic Cosolvent Resistance. Angew. Chem. Int. Ed. 2021, 60, 11448–11456. [Google Scholar] [CrossRef]
- Bertelli, A.; D’Ursi, P.; Campisi, G.; Messali, S.; Milanesi, M.; Giovanetti, M.; Ciccozzi, M.; Caccuri, F.; Caruso, A. Role of Q675H Mutation in Improving SARS-CoV-2 Spike Interaction with the Furin Binding Pocket. Viruses 2021, 13, 2511. [Google Scholar] [CrossRef]
- Richards, F.M.; Kundrot, C.E. Identification of Structural Motifs from Protein Coordinate Data: Secondary Structure and First-Level Supersecondary Structure. Proteins 1988, 3, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Ghosh, S.; Basu, S.; Bhattacharyya, D.; Datta, A.K.; Banerjee, R. Can the Jigsaw Puzzle Model of Protein Folding Re-assemble a Hydrophobic Core? Proteins 2022, accepted. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.D.; Roy, S. Hydrophobic Basis of Packing in Globular Proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 4643–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, K.; Yamagata, Y.; Yutani, K. A General Rule for the Relationship between Hydrophobic Effect and Conformational Stability of a Protein: Stability and Structure of a Series of Hydrophobic Mutants of Human Lysozyme. J. Mol. Biol. 1998, 280, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. Structural Relaxation in Protein Molecules Studied by Fluorescence Spectroscopy. J. Mol. Struct. 1984, 114, 45–48. [Google Scholar] [CrossRef]
- Ramachandran Plot—An Overview. Science Direct Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ramachandran-plot (accessed on 5 November 2021).
- Heinig, M.; Frishman, D. STRIDE: A Web Server for Secondary Structure Assignment from Known Atomic Coordinates of Proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [Green Version]
- Kleywegt, G.J.; Jones, T.A. Phi/Psi-Chology: Ramachandran Revisited. Structure 1996, 4, 1395–1400. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, G.N.; Sasisekharan, V. Conformation of Polypeptides and Proteins. Adv. Protein Chem. 1968, 23, 283–438. [Google Scholar]
- Zhou, A.Q.; O’Hern, C.S.; Regan, L. Revisiting the Ramachandran Plot from a New Angle. Protein Sci. Publ. Protein Soc. 2011, 20, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Michel Espinoza-Fonseca, L.; Kast, D.; Thomas, D.D. Molecular Dynamics Simulations Reveal a Disorder-to-Order Transition on Phosphorylation of Smooth Muscle Myosin. Biophys. J. 2007, 93, 2083–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt-Bridge | TotC | Framesp | ACI | Pers |
|---|---|---|---|---|
| 294-ASP-S ↔ 349-LYS-F | 1 | 1 | 1.00 | 0.00003 |
| 683-ARG-S ↔ 191-ASP-F | 4 | 4 | 1.00 | 0.00013 |
| 654-GLU-S ↔ 357-ARG-F | 23 | 23 | 1.00 | 0.00077 |
| 683-ARG-S ↔ 264-ASP-F | 196 | 64 | 3.06 | 0.00213 |
| 682-ARG-S ↔ 233-ASP-F | 73 | 73 | 1.00 | 0.00243 |
| 654-GLU-S ↔ 359-LYS-F | 149 | 131 | 1.14 | 0.00437 |
| 683-ARG-S ↔ 230-GLU-F | 247 | 185 | 1.34 | 0.00617 |
| 685-ARG-S ↔ 236-GLU-F | 251 | 148 | 1.70 | 0.00493 |
| 214-ARG-S ↔ 112-GLU-F | 729 | 276 | 2.64 | 0.00920 |
| 627-ASP-S ↔ 359-LYS-F | 1247 | 1173 | 1.06 | 0.03910 |
| 627-ASP-S ↔ 357-ARG-F | 2763 | 1460 | 1.89 | 0.04867 |
| 682-ARG-S ↔ 236-GLU-F | 6374 | 1657 | 3.85 | 0.05523 |
| 685-ARG-S ↔ 264-ASP-F | 16,141 | 7146 | 2.26 | 0.23820 |
| 654-GLU-S ↔ 193-ARG-F | 45,873 | 19,387 | 2.37 | 0.64623 |
| 685-ARG-S ↔ 306-ASP-F | 37,148 | 26,969 | 1.38 | 0.89897 |
| 683-ARG-S ↔ 236-GLU-F | 43,275 | 25,546 | 1.69 | 0.85153 |
| 682-ARG-S ↔ 230-GLU-F | 75,112 | 27,762 | 2.71 | 0.92540 |
| Subjects | <∆Hvdw> | <∆Helec> | <∆Smc> | <∆Ssc> | <∆Gbinding> |
|---|---|---|---|---|---|
| RR1CoV-2 | −21.722 (2.669) | −8.296 (0.871) | 17.890 (2.276) | 23.386 (3.432) | 3.687 (3.868) |
| ZR1CoV-2 | −18.748 (1.871) | −8.617 (0.956) | 16.498 (1.889) | 22.004 (2.243) | 3.043 (3.843) |
| ZR1CoV | −13.566 (1.695) | −5.556 (0.582) | 12.663 (1.746) | 14.218 (1.506) | 6.468 (3.135) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Ghosh, P.; Bandyopadhyay, A.; Basu, S. Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges. Vaccines 2022, 10, 301. https://doi.org/10.3390/vaccines10020301
Roy S, Ghosh P, Bandyopadhyay A, Basu S. Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges. Vaccines. 2022; 10(2):301. https://doi.org/10.3390/vaccines10020301
Chicago/Turabian StyleRoy, Sourav, Prithwi Ghosh, Abhirup Bandyopadhyay, and Sankar Basu. 2022. "Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges" Vaccines 10, no. 2: 301. https://doi.org/10.3390/vaccines10020301