
Inhibition of the Peroxygenase Lytic Polysaccharide Monooxygenase by Carboxylic Acids and Amino Acids

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Enzymes
2.2. Determination of LPMO and H2O2 Concentration
2.3. LPMO Turbidity Assay
2.4. LPMO-Activity Assays Based on 2,6-DMP or Hydrocoerulignone
2.5. PASC Batch-Conversion Experiments
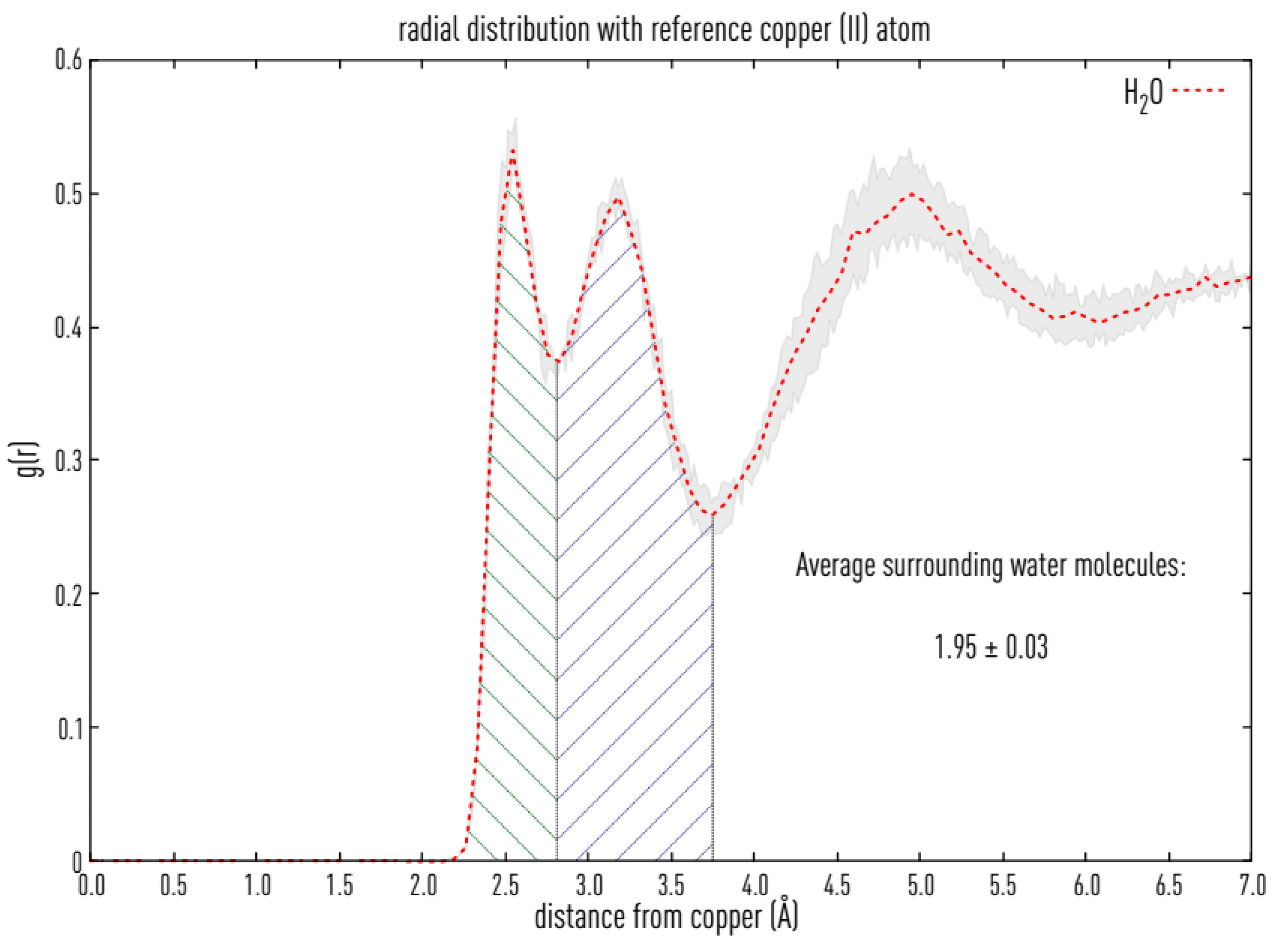
2.6. MD Simulations
2.7. QM Calculations
3. Results
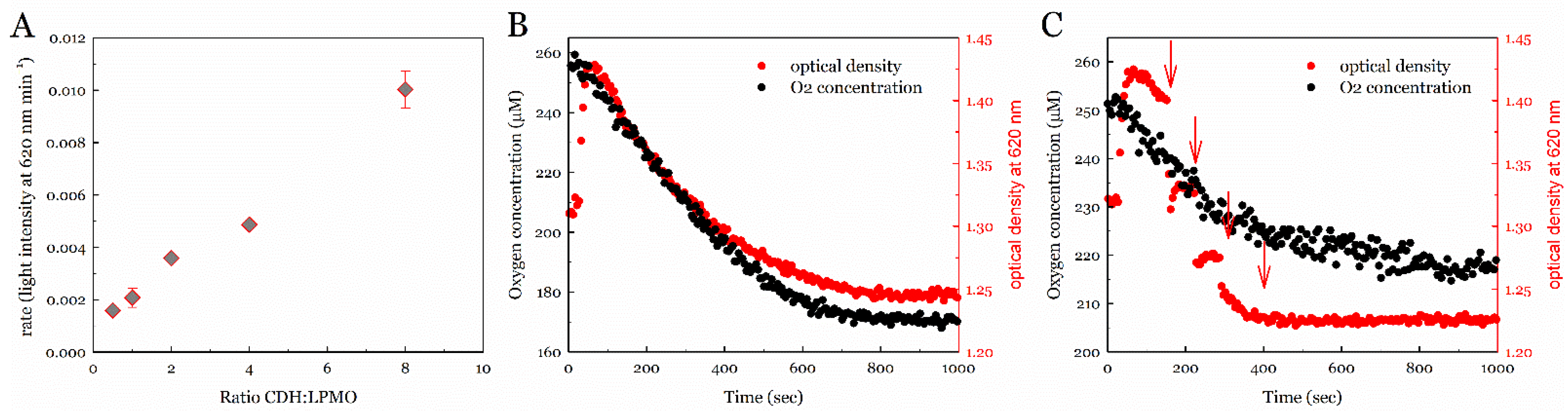
3.1. Assessing the Peroxygenase Reactivity of LPMO
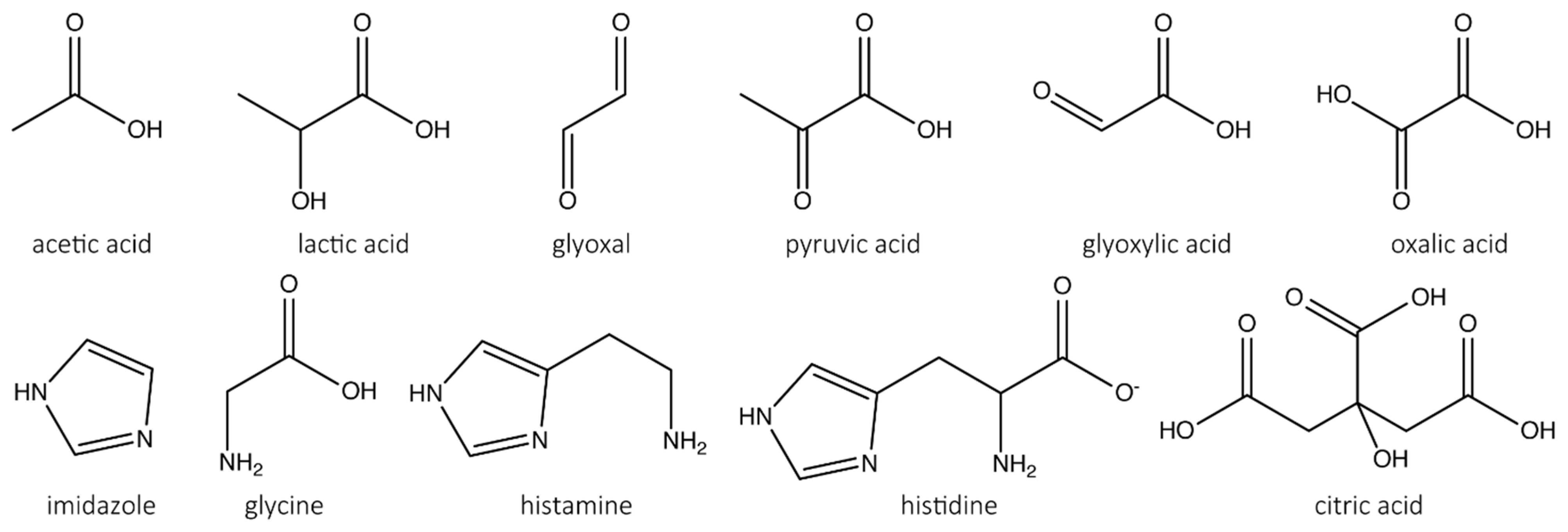
3.2. Screening for LPMO Inhibitors
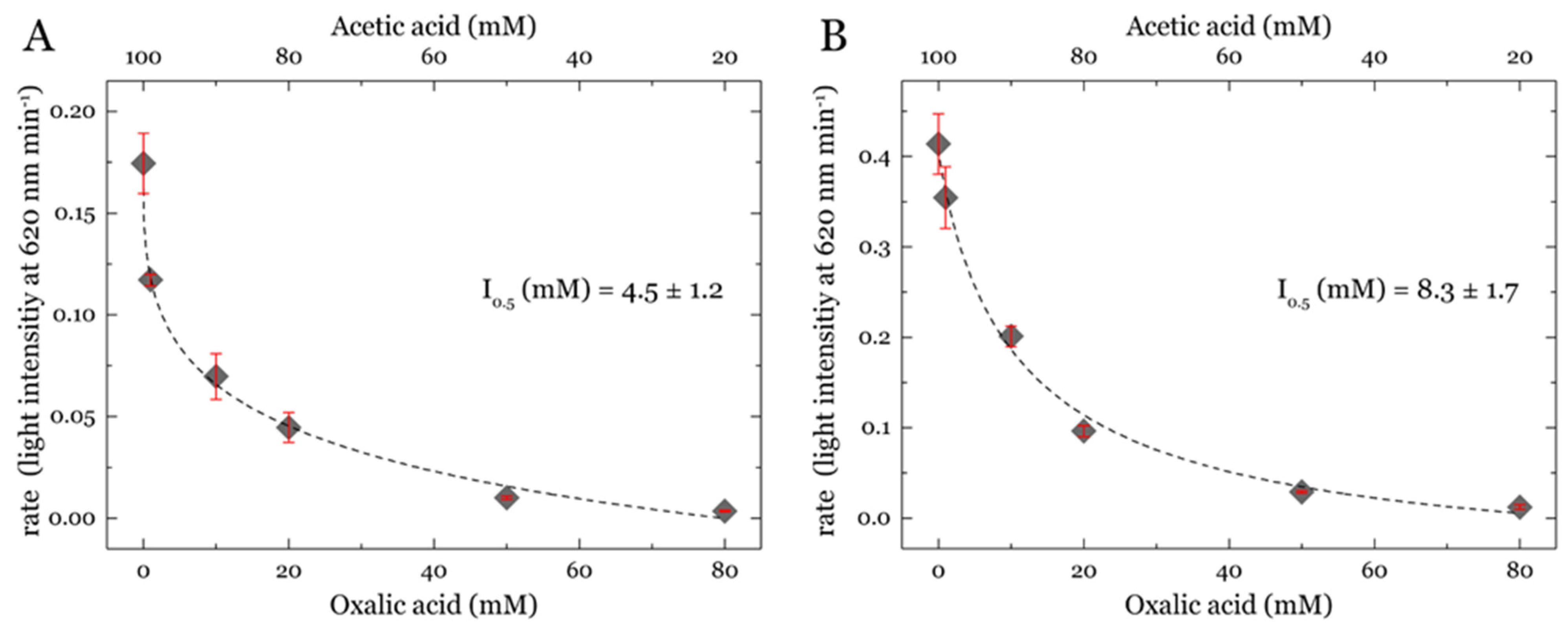
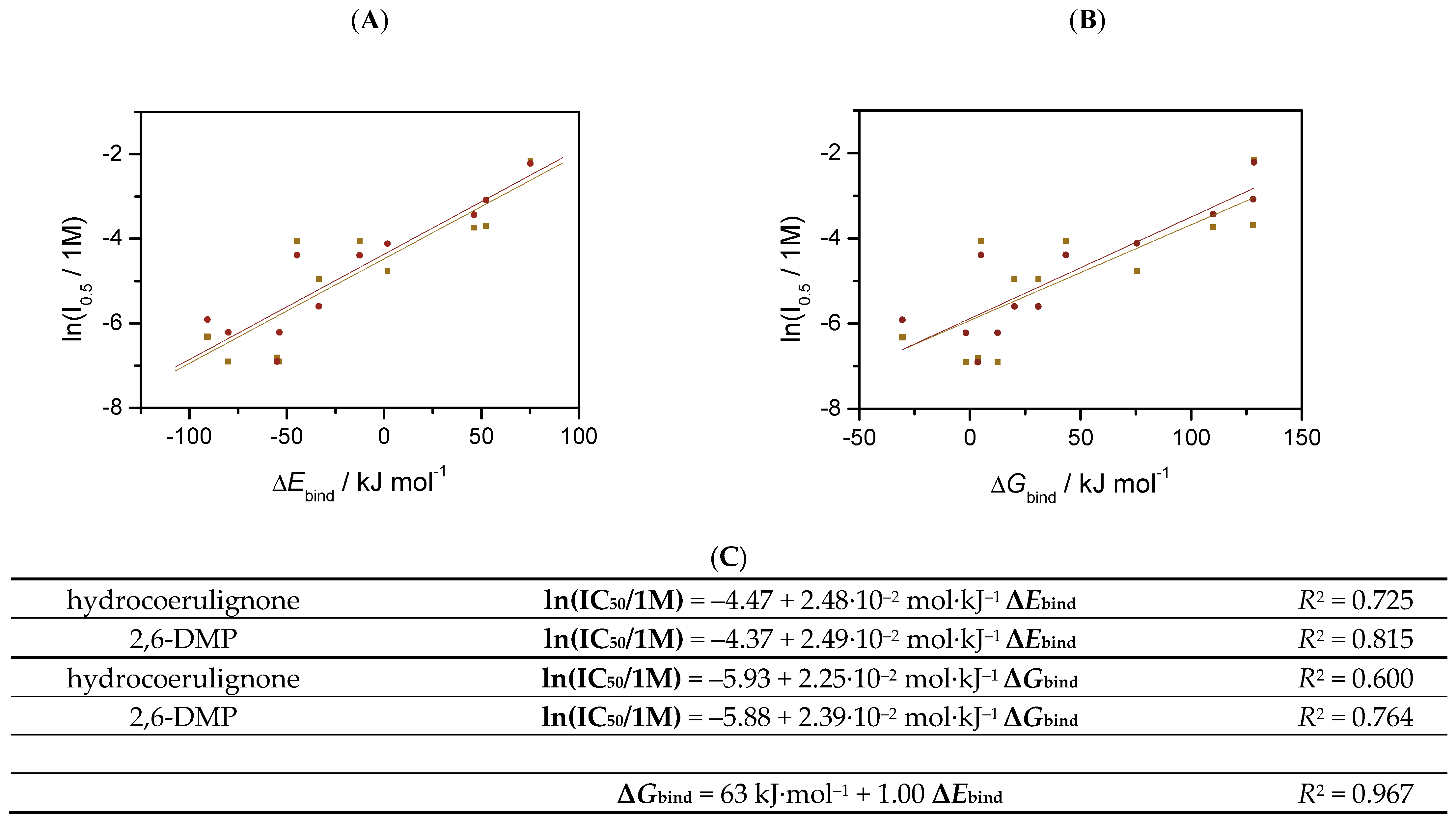
3.3. Determination of the Half-Maximal Inhibitory Concentration (IC50)
3.4. Inhibitory Effect on H2O2 Production by LPMO
3.5. Turbidity Assay for Detecting LPMO Activity on PASC in the Presence of Inhibitors
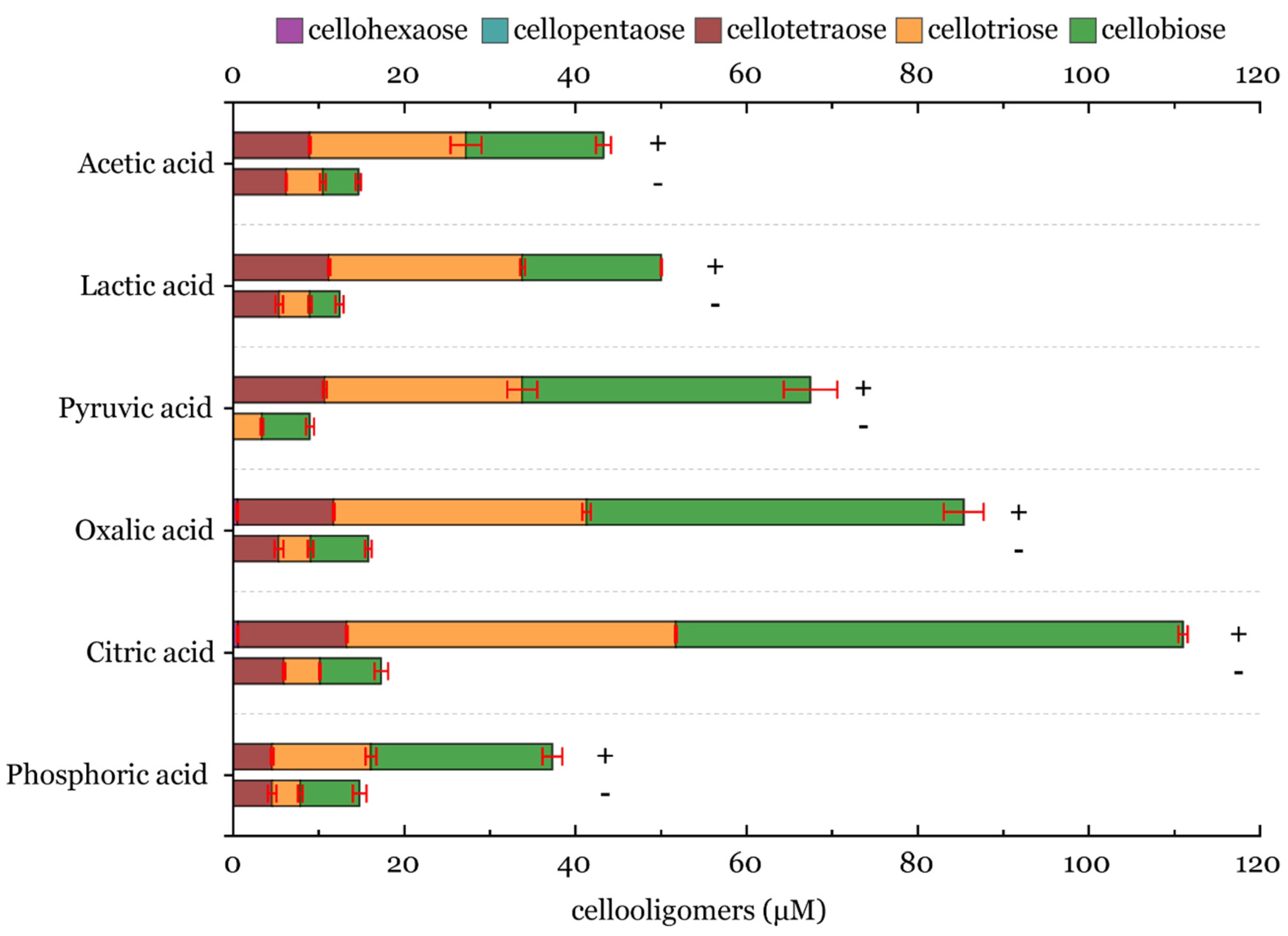
3.6. Batch Conversion of Cellulose with LPMO and Analysis Using HPLC in the Presence of an Inhibitor
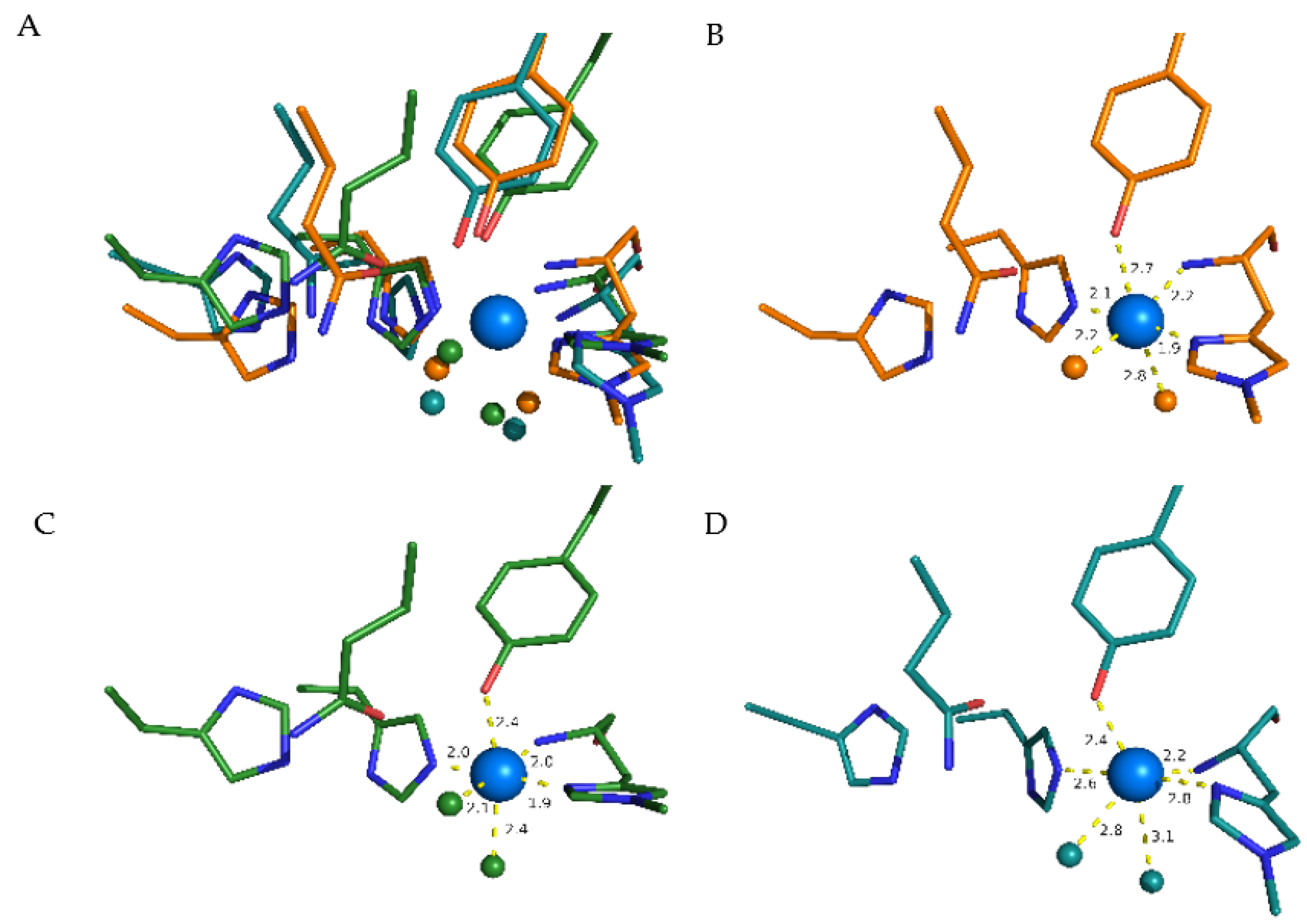
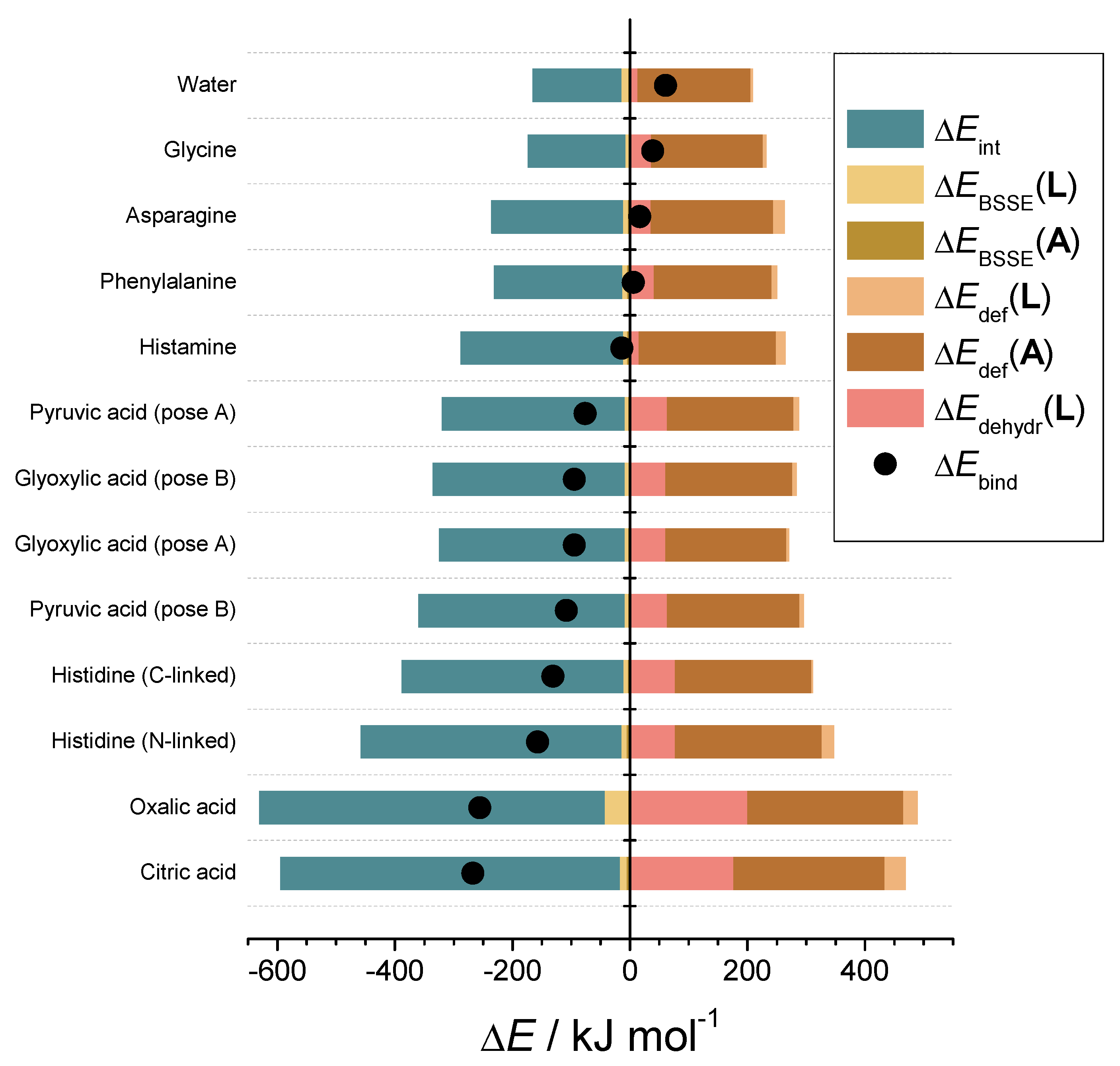
3.7. Structural Causes of LPMO Copper Active Site Inhibition
3.8. Inhibitor–Catalytic Site Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| [2,6-DMP] | 2:6-dimethoxyphenol |
| [hydrocoerulignone] | 3,3′,5,5′-tetramethoxy-4,4′-dihydroxybiphenyl |
| [coerulignone] | 3,3′,5,5′-tetramethoxydiphenoquinone |
| [BSSE] | Basis set superposition error |
| [DEE] | Diethyl ether |
| [DMSO] | Dimethylsulfoxide |
| [H2O2] | Hydrogen peroxide |
| [LPMO] | Lytic polysaccharide monooxygenase |
| [MF] | Molecular dynamics |
| [NBO] | Natural bond orbital |
| [NPA] | Natural population analysis |
| [PASC] | Phosphoric-acid-swollen cellulose |
| [QM] | Quantum mechanical |
| [NaOH] | Sodium hydroxide |
| [SD] | Standard deviation |
| [SE] | Standard error |
References
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.C.N.; Johansen, K.S.; Krogh, K.B.R.M.; Jørgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme That Exploits Biomass Components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsberg, Z.; Vaaje-Kolstad, G.; Westereng, B.; Bunsæ, A.C.; Stenstrøm, Y.; Mackenzie, A.; Sørlie, M.; Horn, S.J.; Eijsink, V.G.H. Cleavage of Cellulose by a Cbm33 Protein. Protein Sci. 2011, 20, 1479–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Hangasky, J.A.; Detomasi, T.C.; Marletta, M.A. Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases. Trends Chem. 2019, 1, 198–209. [Google Scholar] [CrossRef]
- Rieder, L.; Stepnov, A.A.; Sørlie, M.; Eijsink, V.G.H. Fast and Specific Peroxygenase Reactions Catalyzed by Fungal Mono-Copper Enzymes. Biochemistry 2021, 60, 3633–3643. [Google Scholar] [CrossRef]
- Kuusk, S.; Bissaro, B.; Kuusk, P.; Forsberg, Z.; Eijsink, V.G.H.; Sørlie, M.; Valjamae, P. Kinetics of H2O2-Driven Degradation of Chitin by a Bacterial Lytic Polysaccharide Monooxygenase. J. Biol. Chem. 2018, 293, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef]
- Filandr, F.; Kavan, D.; Kracher, D.; Laurent, C.V.F.P.; Ludwig, R.; Man, P.; Halada, P. Structural Dynamics of Lytic Polysaccharide Monooxygenase during Catalysis. Biomolecules 2020, 10, 242. [Google Scholar] [CrossRef] [Green Version]
- Paradisi, A.; Johnston, E.M.; Tovborg, M.; Nicoll, C.R.; Ciano, L.; Dowle, A.; Mcmaster, J.; Hancock, Y.; Davies, G.J.; Walton, P.H. Formation of a Copper(II)-Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by H2O2. J. Am. Chem. Soc. 2019, 141, 18585–18599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommhagen, M.; Westphal, A.H.; Hilgers, R.; Koetsier, M.J.; Hinz, S.W.A.; Visser, J.; Gruppen, H.; van Berkel, W.J.H.; Kabel, M.A. Quantification of the Catalytic Performance of C1-Cellulose-Specific Lytic Polysaccharide Monooxygenases. Appl. Microbiol. Biotechnol. 2018, 102, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Hegnar, O.A.; Petrovic, D.M.; Bissaro, B.; Alfredsen, G.; Várnai, A.; Eijsink, V.G.H. PH-Dependent Relationship between Catalytic Activity and Hydrogen Peroxide Production Shown via Characterization of a Lytic Polysaccharide Monooxygenase from Gloeophyllum trabeum. Appl. Environ. Microbiol. 2019, 85, e02612-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmayr, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular Electron Transfer Systems Fuel Cellulose Oxidative Degradation. Science 2016, 352, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Eijsink, V.G.H.; Petrovic, D.; Forsberg, Z.; Mekasha, S.; Røhr, Å.K.; Várnai, A.; Bissaro, B.; Vaaje-Kolstad, G. On the Functional Characterization of Lytic Polysaccharide Monooxygenases (LPMOs). Biotechnol. Biofuels 2019, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslmayr, E.; Hanžek, M.; Hanrahan, A.; Leitner, C.; Kittl, R.; Šantek, B.; Oostenbrink, C.; Ludwig, R. A Fast and Sensitive Activity Assay for Lytic Polysaccharide Monooxygenase. Biotechnol. Biofuels 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, K.E.H.; Simmons, T.J.; Dupree, P.; Poulsen, J.-C.N.; Hemsworth, G.R.; Ciano, L.; Johnston, E.M.; Tovborg, M.; Johansen, K.S.; von Freiesleben, P.; et al. The Molecular Basis of Polysaccharide Cleavage by Lytic Polysaccharide Monooxygenases. Nat. Chem. Biol. 2016, 12, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Agger, J.W.; Isaksen, T.; Varnai, A.; Vidal-Melgosa, S.; Willats, W.G.T.; Ludwig, R.; Horn, S.J.; Eijsink, V.G.H.; Westereng, B. Discovery of LPMO Activity on Hemicelluloses Shows the Importance of Oxidative Processes in Plant Cell Wall Degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 6287–6292. [Google Scholar] [CrossRef] [Green Version]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. A C4-Oxidizing Lytic Polysaccharide Monooxygenase Cleaving Both Cellulose and Cello-Oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [Green Version]
- Loose, J.S.M.; Forsberg, Z.; Fraaije, M.W.; Eijsink, V.G.H.; Vaaje-Kolstad, G. A Rapid Quantitative Activity Assay Shows That the Vibrio cholerae Colonization Factor GbpA Is an Active Lytic Polysaccharide Monooxygenase. FEBS Lett. 2014, 588, 3435–3440. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Chylenski, P.; Bissaro, B.; Eijsink, V.G.H.; Horn, S.J. The Impact of Hydrogen Peroxide Supply on LPMO Activity and Overall Saccharification Efficiency of a Commercial Cellulase Cocktail. Biotechnol. Biofuels 2018, 11, 209. [Google Scholar] [CrossRef]
- Tokin, R.; Ipsen, J.Ø.; Poojary, M.M.; Jensen, P.E.; Olsson, L.; Johansen, K.S. Inhibition of Lpmos by Fermented Persimmon Juice. Biomolecules 2021, 11, 1890. [Google Scholar] [CrossRef]
- Tokin, R.; Frandsen, K.E.H.; Ipsen, J.Ø.; Lo Leggio, L.; Poojary, M.M.; Berrin, J.G.; Grisel, S.; Brander, S.; Jensen, P.E.; Johansen, K.S. Inhibition of Lytic Polysaccharide Monooxygenase by Natural Plant Extracts. New Phytol. 2021, 232, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Andlar, M.; Furtmüller, P.G.; Ludwig, R. Active-Site Copper Reduction Promotes Substrate Binding of Fungal Lytic Polysaccharide Monooxygenase and Reduces Stability. J. Biol. Chem. 2018, 293, 1676–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenelli, L.; Squina, F.M.; Felby, C.; Cannella, D. Laccase-Derived Lignin Compounds Boost Cellulose Oxidative Enzymes AA9. Biotechnol. Biofuels 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, S.; Kont, R.; Kuusk, P.; Heering, A.; Sørlie, M.; Bissaro, B.; Eijsink, V.G.H.; Väljamäe, P. Kinetic Insights into the Role of the Reductant in H2O2-Driven Degradation of Chitin by a Bacterial Lytic Polysaccharide Monooxygenase. J. Biol. Chem. 2019, 294, 1516–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusakov, A.V.; Bulakhov, A.G.; Demin, I.N.; Sinitsyn, A.P. Monitoring of Reactions Catalyzed by Lytic Polysaccharide Monooxygenases Using Highly-Sensitive Fluorimetric Assay of the Oxygen Consumption Rate. Carbohydr. Res. 2017, 452, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kittl, R.; Kracher, D.; Burgstaller, D.; Haltrich, D.; Ludwig, R. Production of Four Neurospora crassa Lytic Polysaccharide Monooxygenases in Pichia pastoris Monitored by a Fluorimetric Assay. Biotechnol. Biofuels 2012, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Sygmund, C.; Kracher, D.; Scheiblbrandner, S.; Zahma, K.; Felice, A.K.G.; Harreither, W.; Kittl, R.; Ludwig, R. Characterization of the Two Neurospora crassa Cellobiose Dehydrogenases and Their Connection to Oxidative Cellulose Degradation. Appl. Environ. Microbiol. 2012, 78, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; Van Gunsteren, W.F. Architecture, Implementation and Parallelisation of the GROMOS Software for Biomolecular Simulation. Comput. Phys. Commun. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Laurent, C.V.F.P.; Breslmayr, E.; Tunega, D.; Ludwig, R.; Oostenbrink, C. Interaction between Cellobiose Dehydrogenase and Lytic Polysaccharide Monooxygenase. Biochemistry 2019, 58, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Ditchfield, K.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* Basis Set for Atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revis. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Koca, J.; Jirouskova, Z.; Svobodova Varekova, R.; Vanek, J. Electronegativity Equalization Method: Parameterization and Validation for Organic Molecules Using the Merz-Kollman-Singh Charge Distribution Scheme. J. Comput. Chem. 2009, 30, 1174–1178. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Filandr, F.; Man, P.; Halada, P.; Chang, H.; Ludwig, R.; Kracher, D. The H2O2-Dependent Activity of a Fungal Lytic Polysaccharide Monooxygenase Investigated with a Turbidimetric Assay. Biotechnol. Biofuels 2020, 13. [Google Scholar] [CrossRef]
- Hansson, H.; Karkehabadi, S.; Mikkelsen, N.; Douglas, N.R.; Kim, S.; Lam, A.; Kaper, T.; Kelemen, B.; Meier, K.K.; Jones, S.M.; et al. High-Resolution Structure of a Lytic Polysaccharide Monooxygenase from Hypocrea jecorina Reveals a Predicted Linker as an Integral Part of the Catalytic Domain. J. Biol. Chem. 2017, 292, 19099–19109. [Google Scholar] [CrossRef] [Green Version]
- Hangasky, J.A.; Iavarone, A.T.; Marletta, M.A. Reactivity of O2 versus H2O2 with Polysaccharide Monooxygenases. Proc. Natl. Acad. Sci. USA 2018, 115, 4915–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aachmann, F.L.; Sorlie, M.; Skjak-Braek, G.; Eijsink, V.G.H.; Vaaje-Kolstad, G. NMR Structure of a Lytic Polysaccharide Monooxygenase Provides Insight into Copper Binding, Protein Dynamics, and Substrate Interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 18779–18784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Beeson IV, W.T.; Phillips, C.M.M.; Marletta, M.A.A.; Cate, J.H.D.H.D.; Beeson, W.T.; Phillips, C.M.M.; Marletta, M.A.A.; Cate, J.H.D.H.D. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure 2012, 20, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracher, D.; Forsberg, Z.; Bissaro, B.; Gangl, S.; Preims, M.; Sygmund, C.; Eijsink, V.G.H.; Ludwig, R. Polysaccharide Oxidation by Lytic Polysaccharide Monooxygenase Is Enhanced by Engineered Cellobiose Dehydrogenase. FEBS J. 2020, 287, 897–908. [Google Scholar] [CrossRef]
- Breslmayr, E.; Laurent, C.V.F.P.; Scheiblbrandner, S.; Jerkovic, A.; Heyes, D.J.; Oostenbrink, C.; Ludwig, R.; Hedison, T.M.; Scrutton, N.S.; Kracher, D. Protein Conformational Change Is Essential for Reductive Activation of Lytic Polysaccharide Monooxygenase by Cellobiose Dehydrogenase. ACS Catal. 2020, 10, 4842–4853. [Google Scholar] [CrossRef]
- Kont, R.; Bissaro, B.; Eijsink, V.G.H.; Väljamäe, P. Kinetic Insights into the Peroxygenase Activity of Cellulose-Active Lytic Polysaccharide Monooxygenases (LPMOs). Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Hedison, T.M.; Breslmayr, E.; Shanmugam, M.; Karnpakdee, K.; Heyes, D.J.; Green, A.P.; Ludwig, R.; Scrutton, N.S.; Kracher, D. Insights into the H2O2-Driven Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases. FEBS J. 2021, 288, 4115–4128. [Google Scholar] [CrossRef]
- Sørensen, P.E.; Bruhn, K.; Lindeløv, F.; Østvold, T.; Bjørseth, A.; Powell, D.L. Kinetics and Equilibria for the Reversible Hydration of the Aldehyde Group in Glyoxylic Acid. Acta Chem. Scand. 1974, 28, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Redington, R.L.; Liang, C.K.J. Vibrational Spectra of Glyoxylic Acid Monomers. J. Mol. Spectrosc. 1984, 104, 25–39. [Google Scholar] [CrossRef]
- Mattioda, G.; Christidis, Y. Glyoxylic Acid. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Lopalco, A.; Douglas, J.; Denora, N.; Stella, V.J. Determination of pKa and Hydration Constants for a Series of α-Keto-Carboxylic Acids Using Nuclear Magnetic Resonance Spectrometry. J. Pharm. Sci. 2016, 105, 664–672. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes a | Ascorbic Acid (mM) | Substrate b | Cosubstrate c | Buffer Species | Conc. (mM) | pH | Temp. (°C) | Reference |
|---|---|---|---|---|---|---|---|---|
| LsAA9A | 4 | PASC, cellooligomers | O2 | ammonium-formate | 100 | 6.0 | 20 | [15] |
| NcAA9C-CBM1 | 1 | XG, PASC, cellooligomers | O2 | sodium-acetate | 10 | 6.0 | 40 | [16] |
| 1 | PASC | O2 | potassium-phosphate | 50 | 6.0 | 30 | [12] | |
| 2 | PASC | O2 | ammonium-acetate | 5 | 6.0 | 50 | [17] | |
| MtAA9B, D | 1 | RAC | O2 | ammonium-acetate | 50 | 5.0 | 50 | [10] |
| sodium-citrate/phosphate | 50 | 3.0–8.0 | 40–50 | |||||
| GtAA9B | 1 | PASC | O2 | Bis-/Tris-HCl | 50 | 6–9 | 45 | [11] |
| H2O2 | Bis-Tris-HCl | 6.5 | ||||||
| SmAA10A | 0.2–5 | β-chitin | O2 | Tris-HCl | 20 | 8.0 | 37 | [3] |
| 0.1 | CNW | H2O2 | sodium-acetate | 50 | 6.1 | 25 | [6] | |
| ScAA10C | 1 | Avicel | H2O2 | sodium-phosphate | 50 | 7.0 | 40–50 | [7] |
| VcAA10B-X-Y-CBM73 | 1 | β-chitin nanofibers | O2 | Bis-Tris-HCl | 50 | 6.8 | 37 | [18] |
| Cellic CTec2 | 0.1–10 | Avicel | H2O2 | sodium-acetate | 50 | 5.0 | 50 | [19] |
| Hydro-Coerulignone | 2,6-DMP | Hydro-Coerulignone | 2,6-DMP | |
|---|---|---|---|---|
| Specific activity with 100 mM inhibitor (U g−1) | Residual activity to 100 mM acetate buffer (%) | |||
| Acetic acid | 97.4 ± 7.2 | 17.5 ± 1.5 | 70.8 ± 5.2 | 76.5 ± 6.6 |
| Lactic acid | 67.6 ± 10.2 | 6.4 ± 0.3 | 49.2 ± 7.4 | 28.1 ± 1.3 |
| Glyoxal | 60.5 ± 4.9 | 4.8 ± 0.3 | 43.9 ± 3.6 | 20.8 ± 1.5 |
| Pyruvic acid | 5.6 ± 0.6 | 0.1 ± 0.1 | 4.1 ± 0.4 | 0.1 ± 0.1 |
| Glyoxylic acid | 1.5 ± 0.4 | 0.1 ± 0.1 | 1.1 ± 0.3 | 0.1 ± 0.1 |
| Oxalic acid | 0.8 ± 0.2 | 0.1 ± 0.1 | 0.6 ± 0.1 | 0.1 ± 0.1 |
| Citric acid | 2.0 ± 0.5 | 0.3 ± 0.1 | 1.5 ± 0.4 | 1.3 ± 0.4 |
| Imidazole | 99.4 ± 5.4 | 18.3 ± 1.1 | 72.2 ± 3.9 | 80.0 ± 4.8 |
| Glycine | 67.7 ± 1.6 | 10.1 ± 0.8 | 49.2 ± 1.2 | 44.1 ± 3.5 |
| Histamine | 2.2 ± 0.6 | 0.8 ± 0.3 | 1.6 ± 0.4 | 3.5 ± 1.3 |
| Histidine | 0.5 ± 0.3 | 0.1 ± 0.1 | 0.4 ± 0.2 | 0.1 ± 0.1 |
| Hydrocoerulignone | 2,6-DMP | |
|---|---|---|
| IC50 (mM) | ||
| Oxalic acid | 1.1 ± 0.1 | 1.0 ± 0.1 |
| Glyoxylic acid | 7.1 ± 0.2 | 3.7 ± 0.6 |
| Pyruvic acid | 17.2 ± 1.0 | 12.4 ± 0.8 |
| Citric acid | 1.8 ± 0.3 | 2.7 ± 0.2 |
| Histidine | 1.0 ± 0.1 | 2.0 ± 0.1 |
| Histamine | 8.5 ± 0.4 | 16.3 ± 0.7 |
| Glycine | 115 ± 20 | 109 ± 6 |
| Asparagine | 24.9 ± 0.2 | 45.8 ± 2.8 |
| Phenylalanine | 23.8 ± 1.1 | 32.4 ± 3.7 |
| Na2SO4 | 31.2 ± 4.7 | 32.8 ± 3.2 |
| NaF | 142 ± 39 | 316 ± 17 |
| Rate (Light Intensity at 620 nm min−1) | Residual Activity (%) | |
|---|---|---|
| Acetic acid | 0.14 ± 0.01 | 84 ± 5 |
| Phosphoric acid | 0.107 ± 0.02 | 65 ± 10 |
| Lactic acid | 0.08 ± 0.02 | 47 ± 13 |
| Citric acid | 0.014 ± 0.001 | 8 ± 1 |
| Oxalic acid | 0.002 ± 0.001 | 1.4 ± 0.6 |
| Nim(Me-His1) | Nam(Me-His1) | Nim(His83) | O(Tyr166) | Leq | Lax | |
|---|---|---|---|---|---|---|
| Water | 1.95 | 2.05 | 1.97 | 2.44 | 2.07 | 2.39 |
| Glycine | 1.96 | 2.06 | 1.97 | 2.35 | 2.01 | 2.80 |
| Asparagine | 1.96 | 2.03 | 2.01 | 2.41 | 1.95 | 3.01 |
| Phenylalanine | 1.96 | 2.04 | 1.95 | 2.34 | 2.00 | 3.90 * |
| Histamine | 2.01 | 2.06 | 2.04 | 2.79 | 2.04 | 2.19 |
| Pyruvic acid a | 1.95 | 2.08 | 1.98 | 2.81 | 2.43 | 1.98 |
| Glyoxylic acid b | 1.97 | 2.05 | 1.99 | 2.51 | 1.96 | 2.53 |
| Glyoxylic acid a | 1.96 | 2.07 | 1.97 | 2.36 | 1.96 | - ** |
| Pyruvic acid b | 1.97 | 2.04 | 1.99 | 2.63 | 1.96 | 2.41 |
| Histidine c | 2.01 | 2.06 | 2.04 | 2.83 | 2.00 | 2.20 |
| Histidine d | 1.94 | 2.05 | 1.96 | 2.71 | 1.91 | - ** |
| Oxalic acid | 1.98 | 2.07 | 2.02 | 3.02 | 1.98 | 2.15 |
| Citric acid | 1.98 | 2.06 | 2.01 | 3.36 | 1.95 | 2.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breslmayr, E.; Poliak, P.; Požgajčić, A.; Schindler, R.; Kracher, D.; Oostenbrink, C.; Ludwig, R. Inhibition of the Peroxygenase Lytic Polysaccharide Monooxygenase by Carboxylic Acids and Amino Acids. Antioxidants 2022, 11, 1096. https://doi.org/10.3390/antiox11061096
Breslmayr E, Poliak P, Požgajčić A, Schindler R, Kracher D, Oostenbrink C, Ludwig R. Inhibition of the Peroxygenase Lytic Polysaccharide Monooxygenase by Carboxylic Acids and Amino Acids. Antioxidants. 2022; 11(6):1096. https://doi.org/10.3390/antiox11061096
Chicago/Turabian StyleBreslmayr, Erik, Peter Poliak, Alen Požgajčić, Roman Schindler, Daniel Kracher, Chris Oostenbrink, and Roland Ludwig. 2022. "Inhibition of the Peroxygenase Lytic Polysaccharide Monooxygenase by Carboxylic Acids and Amino Acids" Antioxidants 11, no. 6: 1096. https://doi.org/10.3390/antiox11061096