
Identification of Hub Genes Associated with Breast Cancer Using Integrated Gene Expression Data with Protein-Protein Interaction Network


Abstract
:1. Background
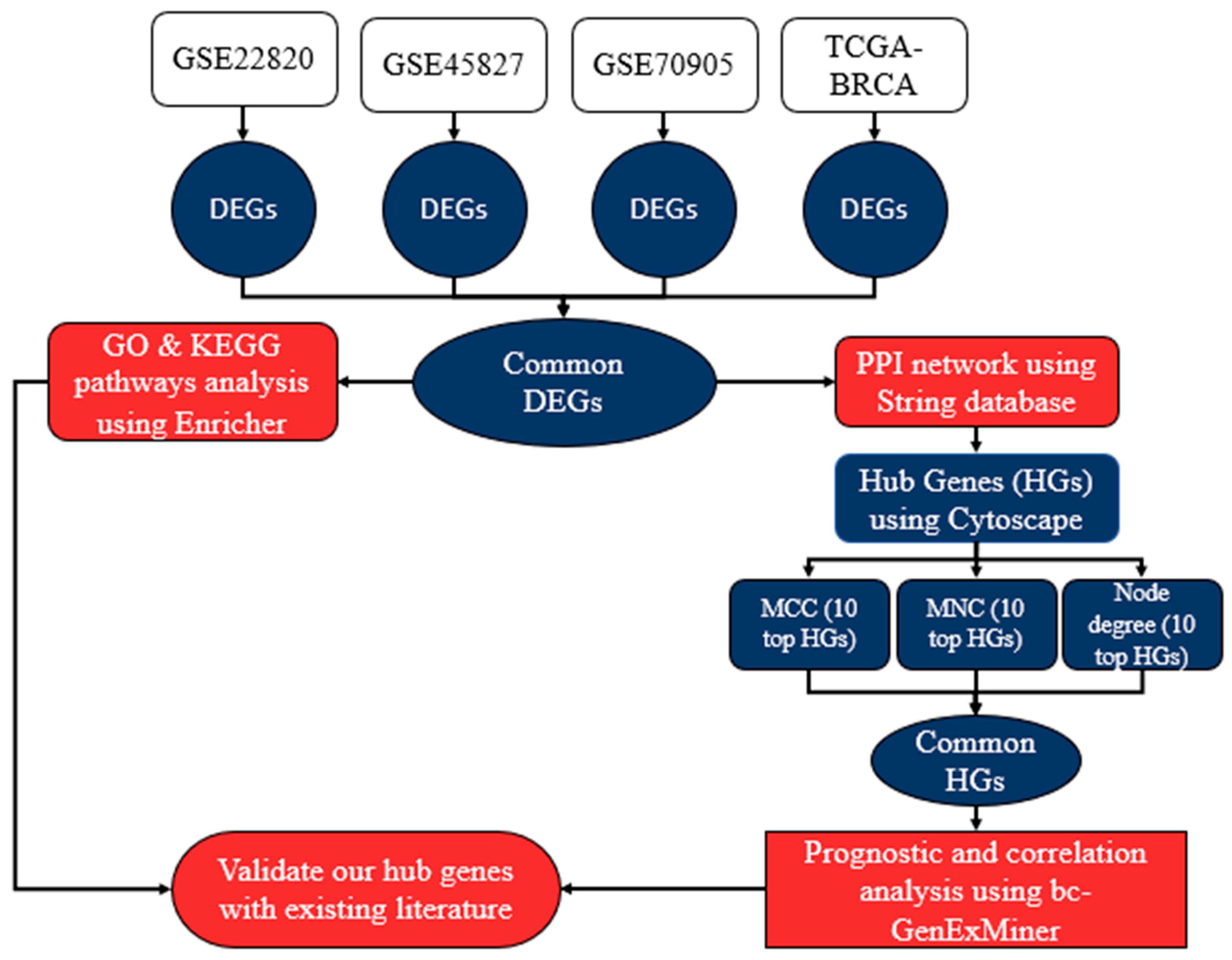
2. Material and Methods
Datasets
3. Differential Gene Expression Analysis
3.1. Microarray Analysis
3.2. RNASeq Analysis
3.3. Gene Ontology (GO) and Gene List Pathway Enrichment Analysis
3.4. Protein-Protein Interaction (PPI) Network Analysis
4. Results
4.1. The GO Term Analysis of the Common DEGs
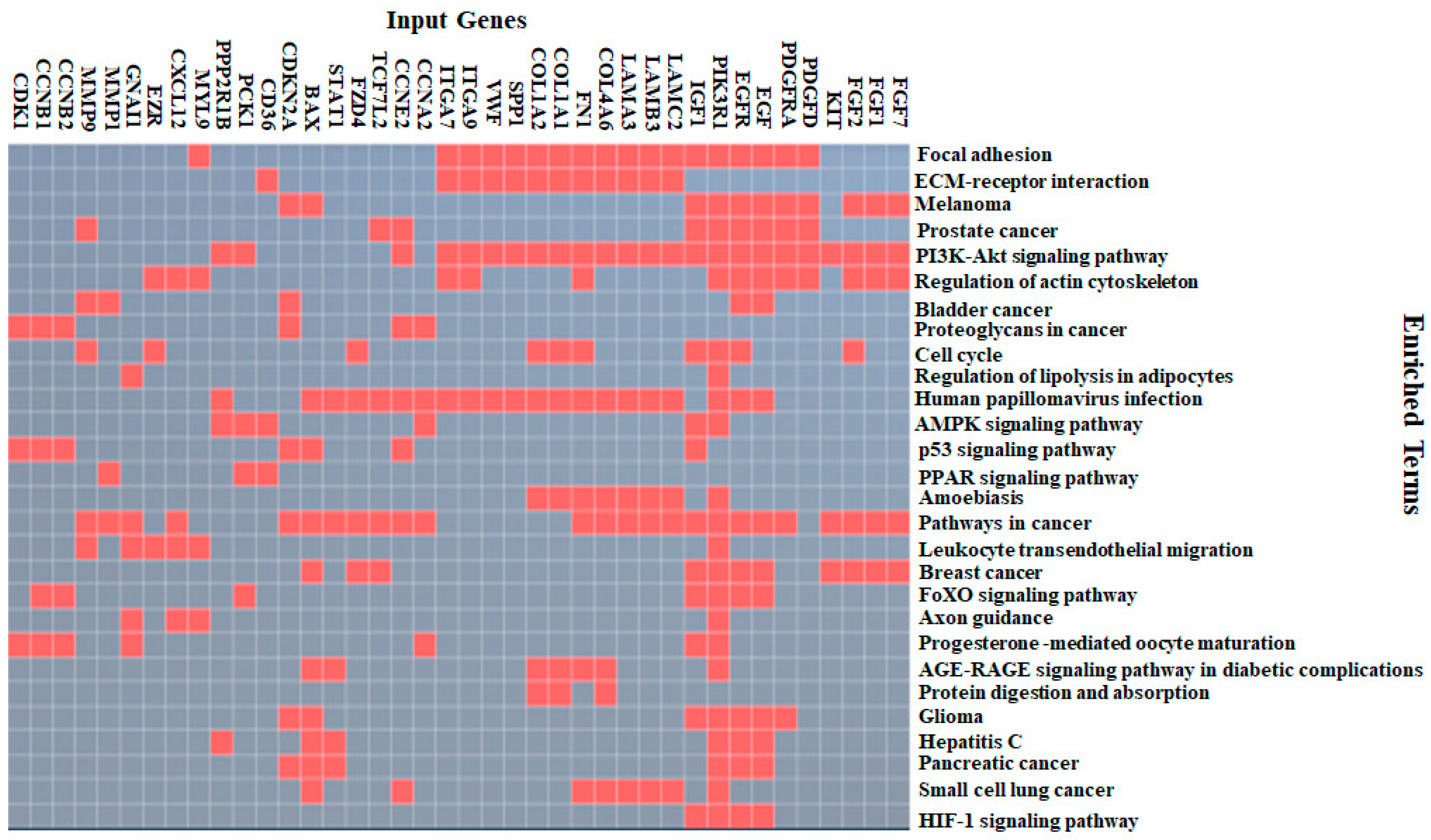
4.2. KEGG Pathway Enrichment Analysis for the DEGs
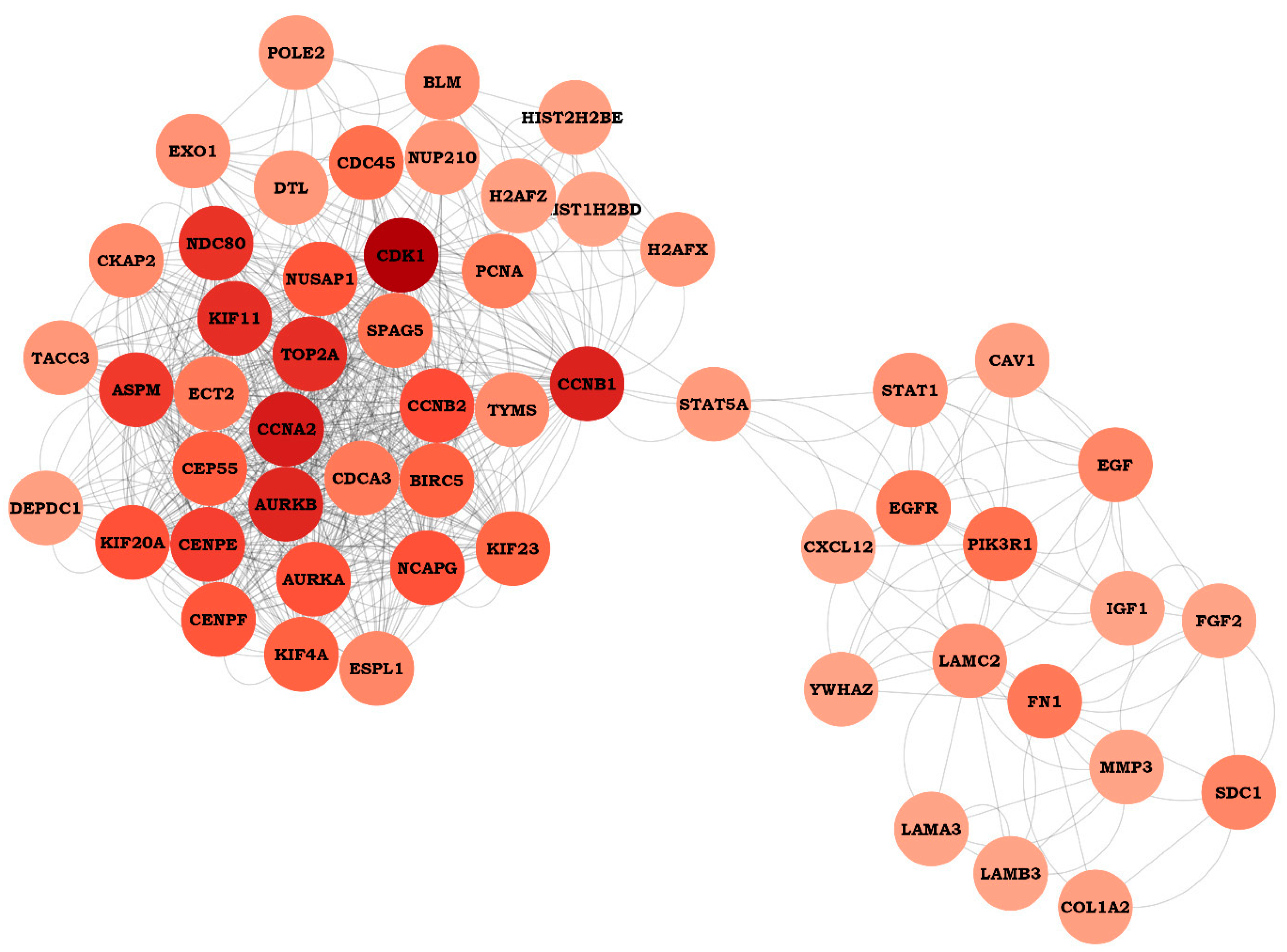
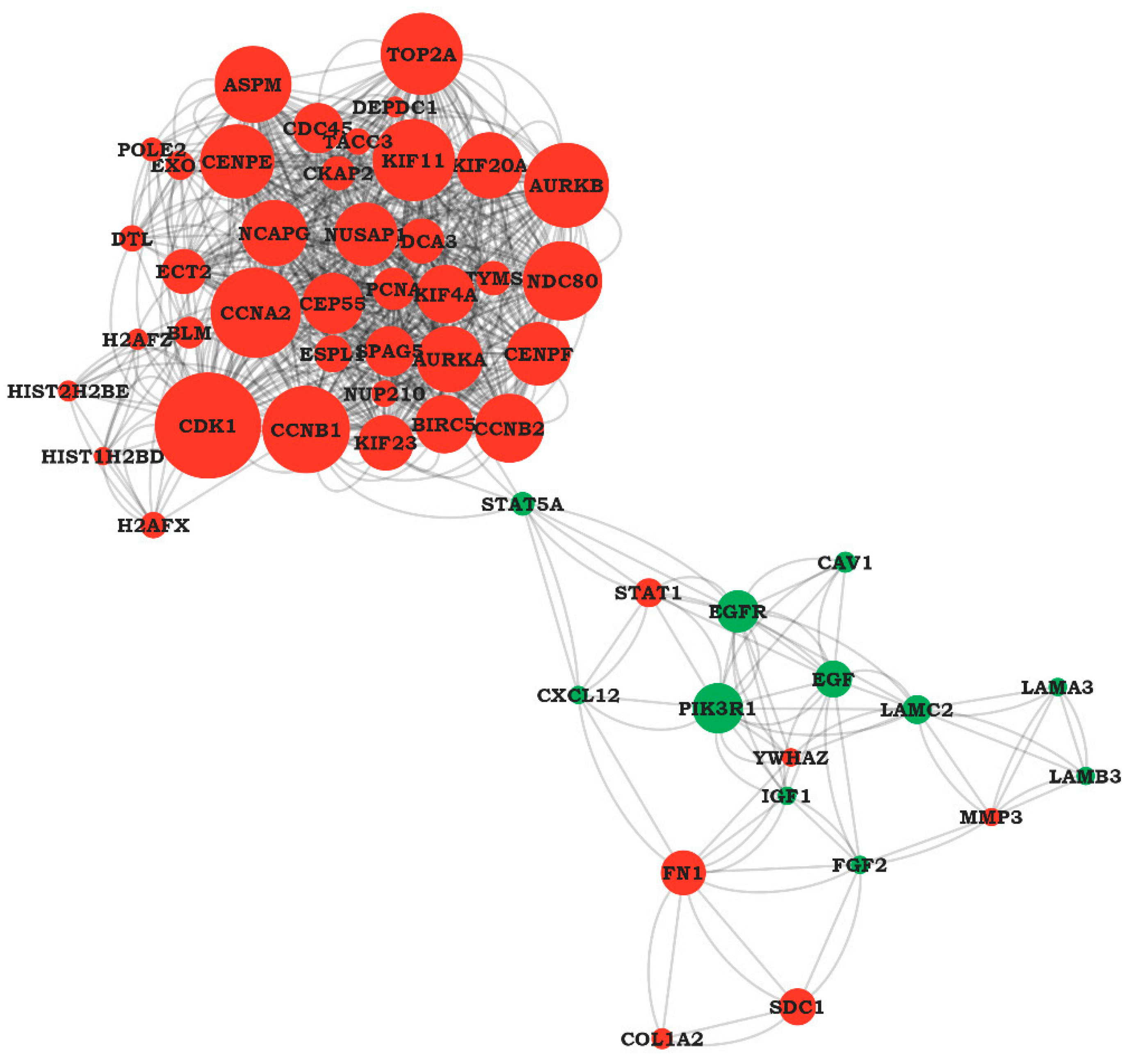
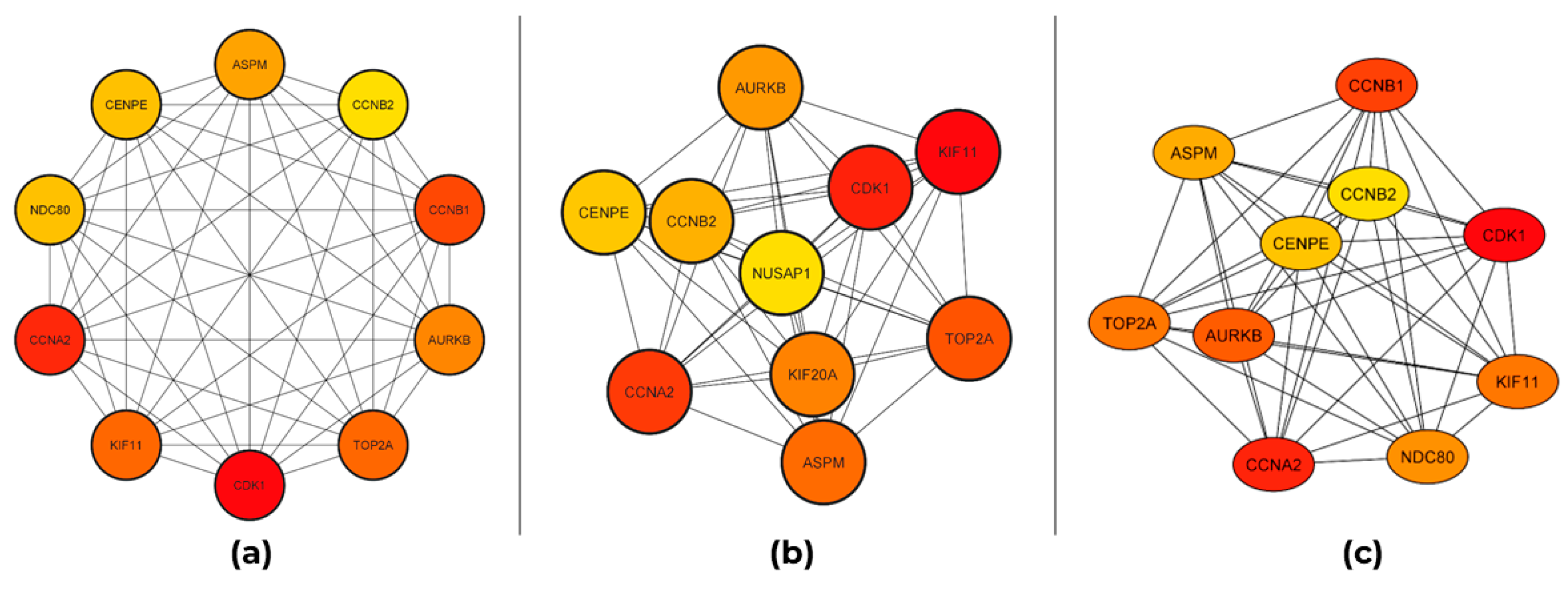
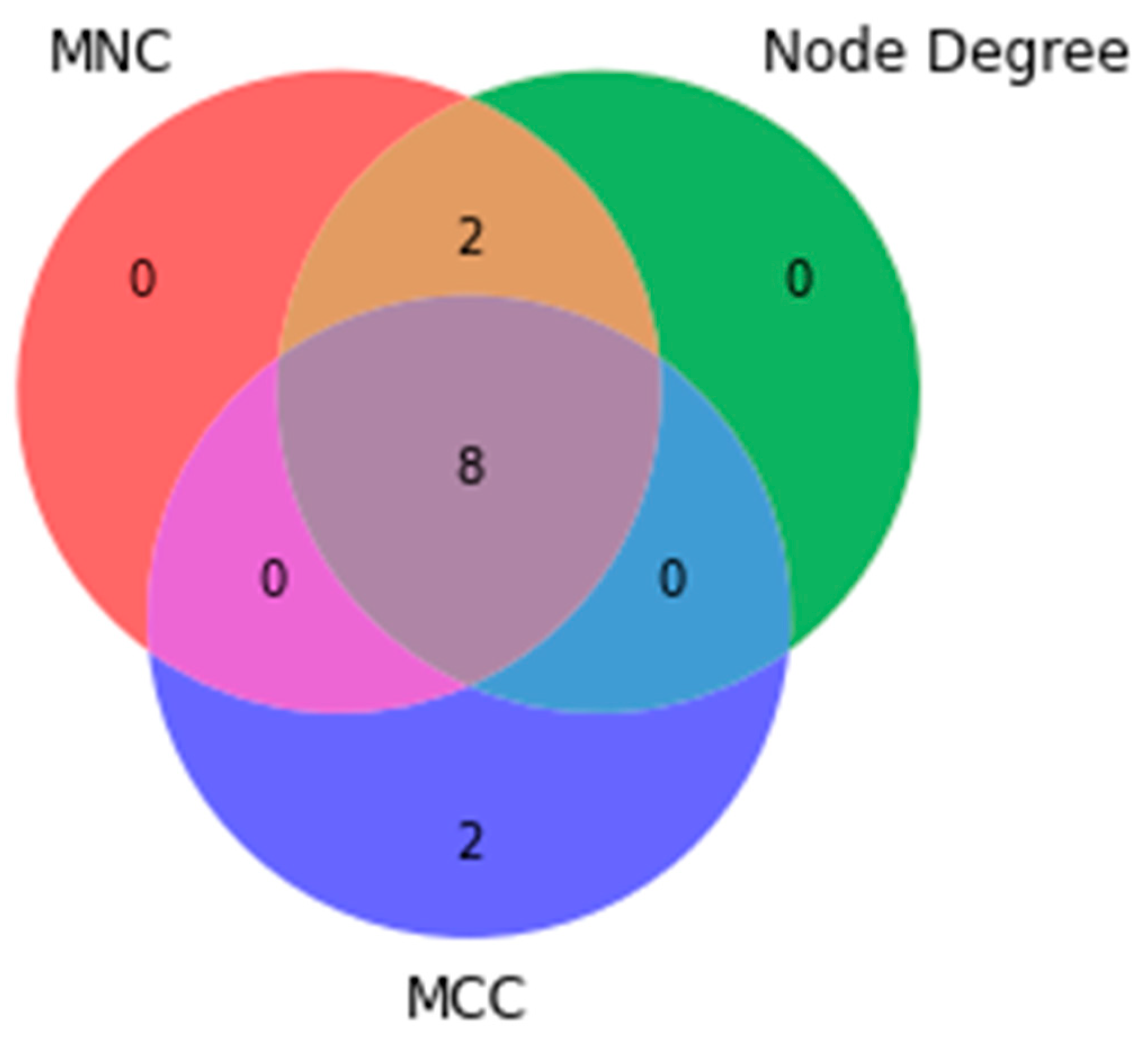
4.3. PPI Network Construction and Hub Genes Selection
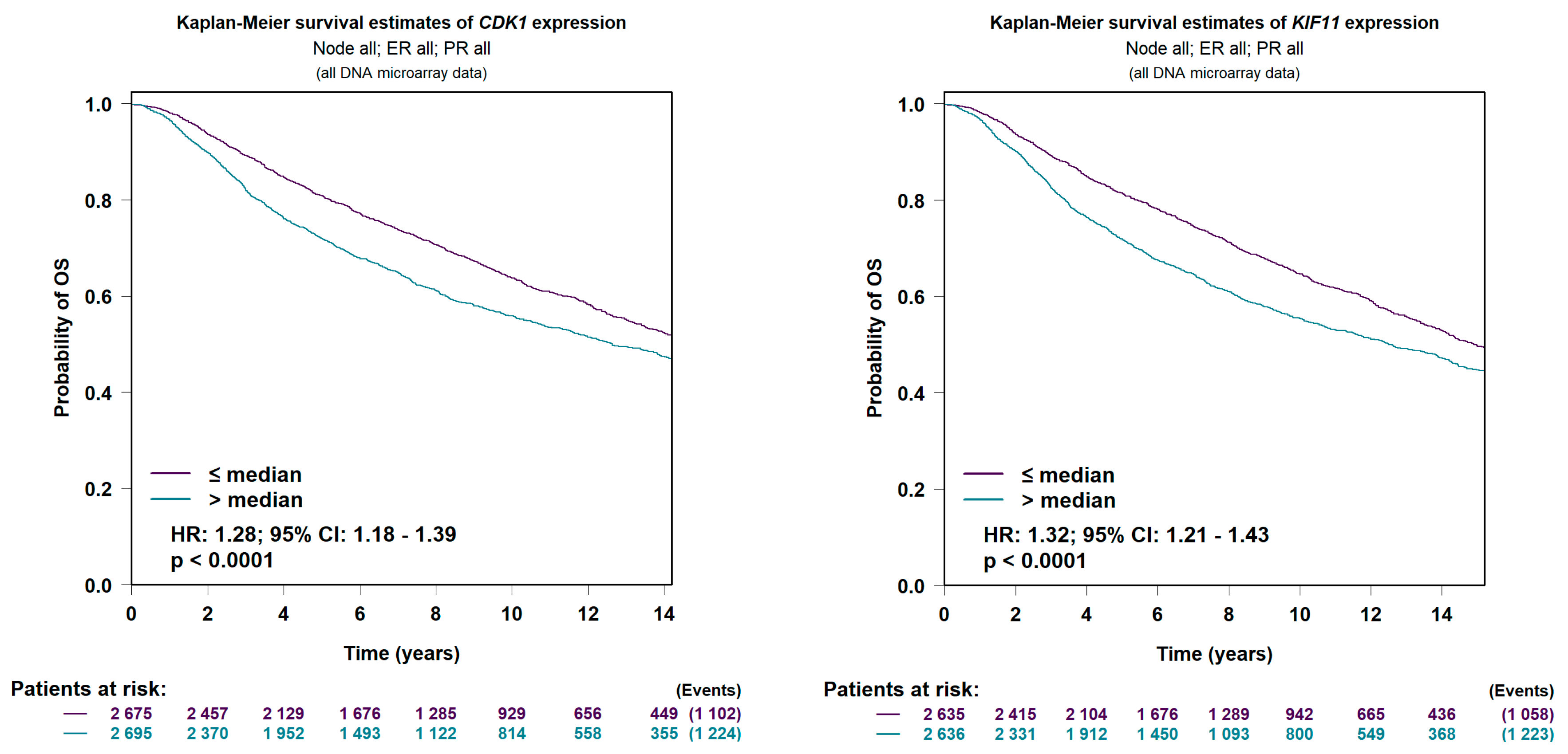
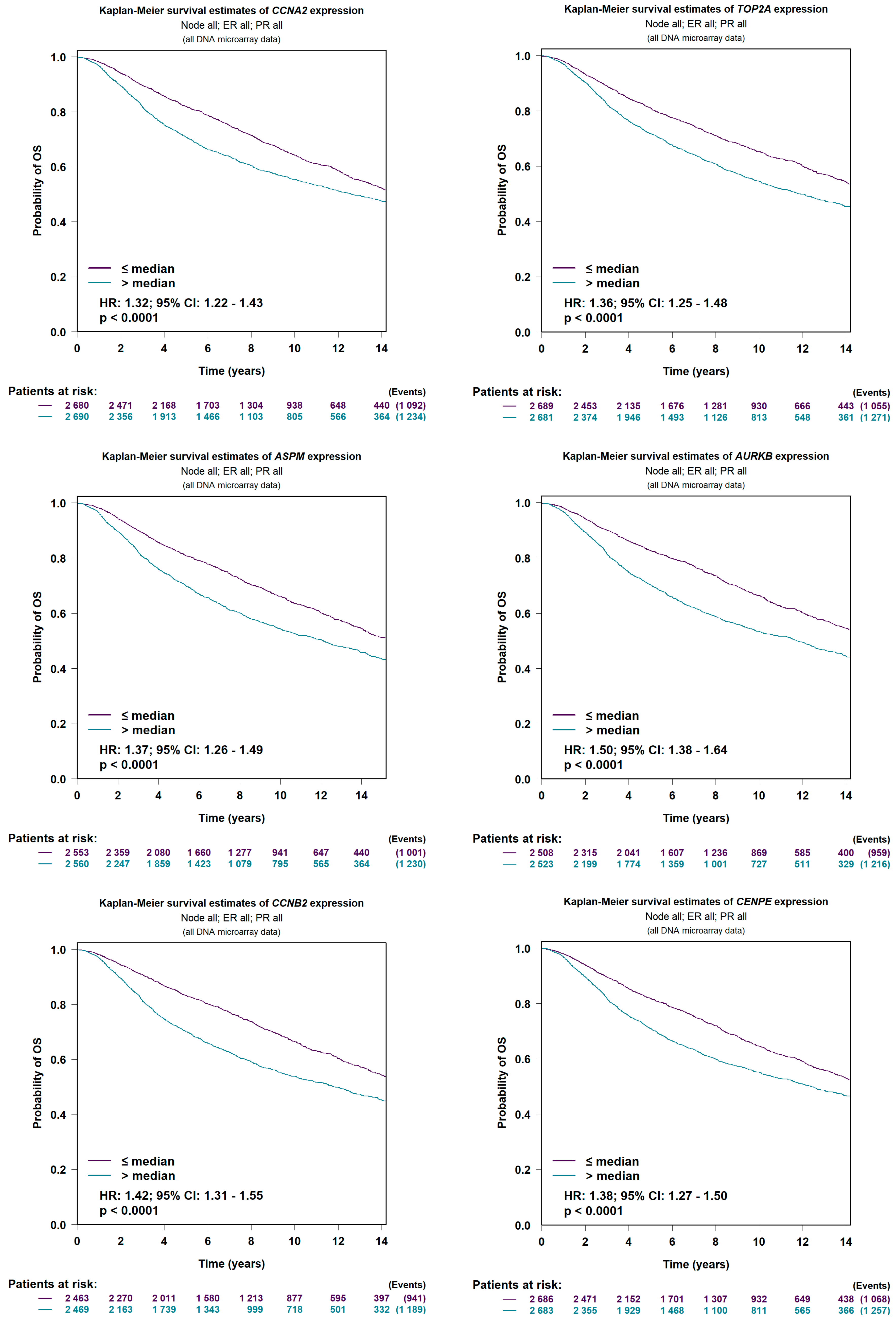
4.4. Prognostic Value Verification of Hub Genes
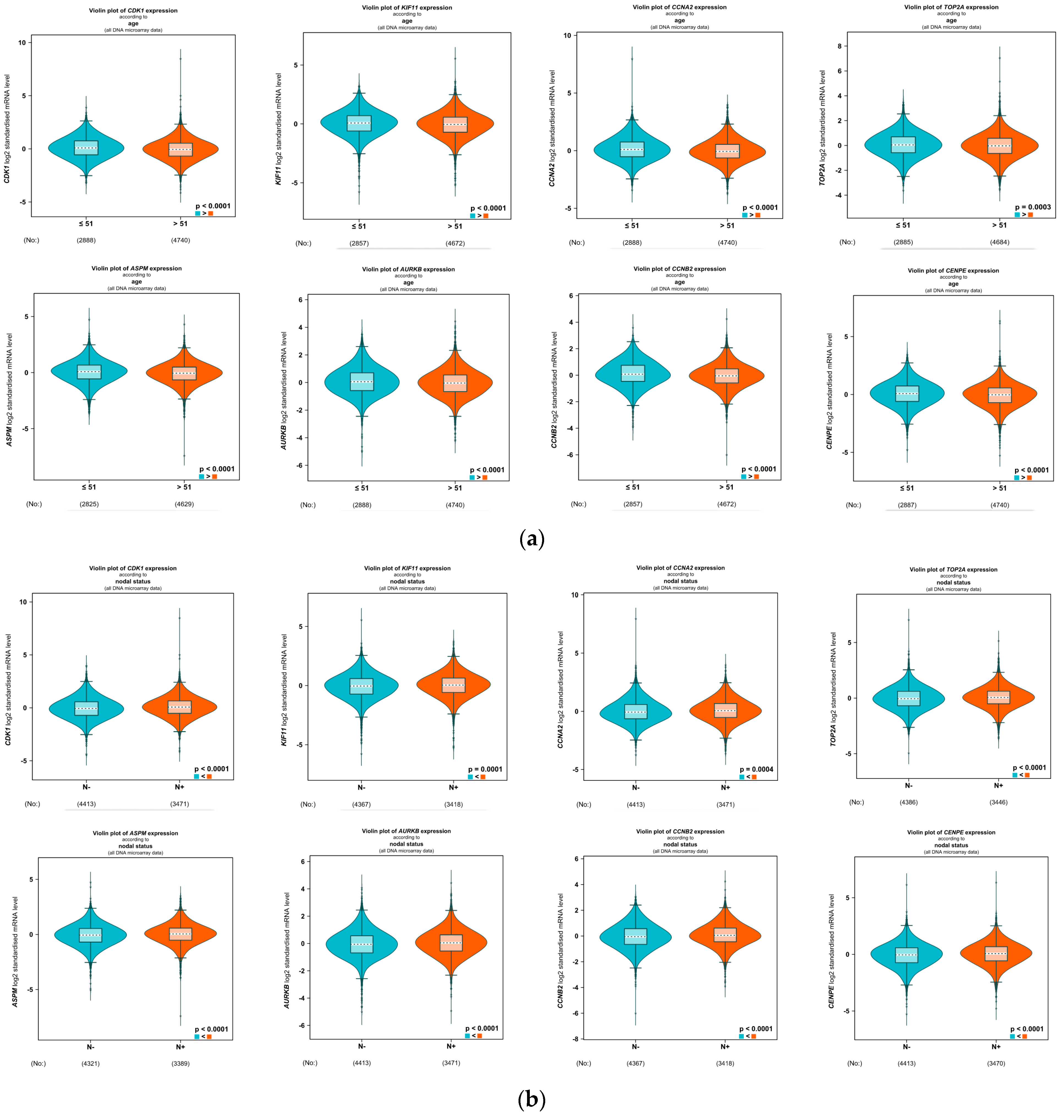
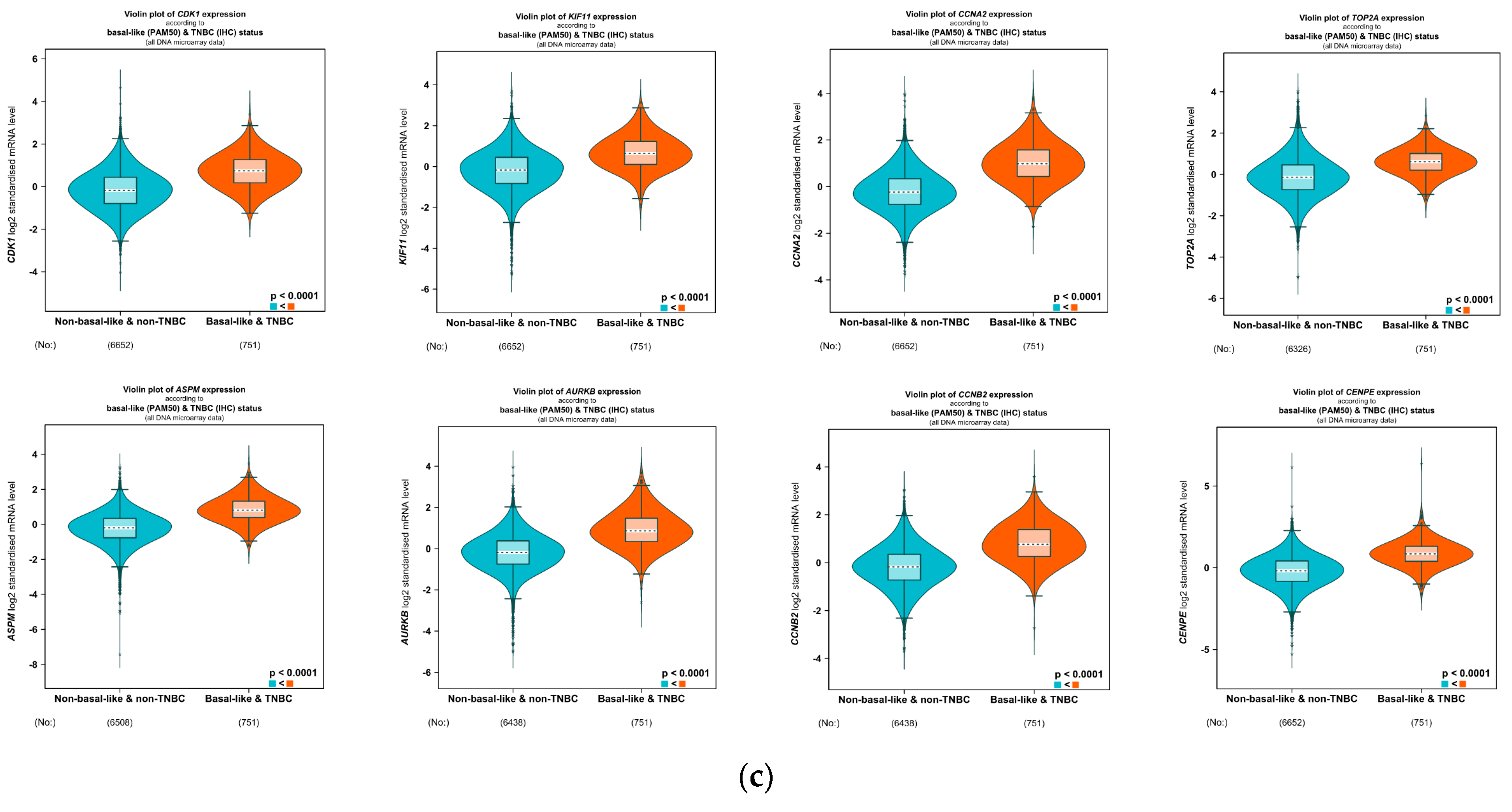
4.5. Correlation Analysis of the Clinicopathological Variables in BC with the Eight Hub Genes
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| DEGs | Differentially Expressed Genes |
| GO | Gene Ontology |
| MCC | Maximal Clique Centrality |
| MCN | Maximum Neighborhood Component |
| NCDs | Non-Communicable Diseases |
| WHO | World Health Organization |
| NGS | Next-Generation Sequencing |
| TNM | Tumour-Node-Metastasis |
| AMD | Age-related Macular Degeneration |
| ECM | ExtraCellular Matrix |
| PPI | Protein-Protein Interaction |
| PaCa | Pancreatic Cancer |
| CRC | Colorectal Cancer |
| KEGG | Kyoto Encyclopaedia of Genes and Genomes |
| PBMC | Peripheral Blood Mononuclear Cell |
| TNF | Tumor Necrosis Factor |
| mRMR | Minimum Redundancy Maximum Relevance |
| limma | Linear Models for Microarrays |
| AAIC | Array-Array Intensity Correlation |
| DE | Differentially Expressed |
References
- WHO. Noncommunicable Diseases 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 30 March 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kumar, R.; Mittal, V.; Gupta, D. Classification models for Invasive Ductal Carcinoma Progression, based on gene expres-sion data-trained supervised machine learning. Sci. Rep. 2020, 10, 4113. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Baehner, F.; Dabbs, D.J.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Breast cancer prognostic classification in the molecular era: The role of histological grade. Breast. Cancer Res. 2010, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Gress, D.M.; Edge, S.B.; Greene, F.L.; Washington, M.K.; Asare, E.A.; Brierley, J.D.; Byrd, D.R.; Compton, C.C.; Jessup, J.M.; Winchester, D.P. Principles of cancer staging. AJCC Cancer Staging Man. 2017, 8, 3–30. [Google Scholar]
- Roberto Cesar, M.-O.; German, L.-B.; Paola Patricia, A.-C.; Eugenia, A.-R.; Elisa Clementina, O.-M.; Jose, C.-O.; Marlon Alberto, P.-M.; Fabio Enrique, M.-P.; Margarita, R.-V. Method based on data mining techniques for breast cancer recurrence analysis. In Proceedings of the International Conference on Swarm Intelligence, Belgrade, Serbia, 14–20 July 2020; Springer: Berlin/Heidelberg, Germany, 2020; pp. 584–596. [Google Scholar]
- Abd-Elnaby, M.; Alfonse, M.; Roushdy, M. Classification of breast cancer using microarray gene expression data: A survey. J. Biomed. Inform. 2021, 117, 103764. [Google Scholar] [CrossRef]
- Ma, J.; Chen, C.; Liu, S.; Ji, J.; Wu, D.; Huang, P.; Wei, D.; Fan, Z.; Ren, L. Identification of a five genes prognosis signature for triple-negative breast cancer using multi-omics methods and bioinformatics analysis. Cancer Gene Ther. 2022, 29, 1578–1589. [Google Scholar] [CrossRef]
- Alam, M.S.; Rahaman, M.M.; Sultana, A.; Wang, G.; Mollah, M.N.H. Statistics and network-based approaches to identify molecular mechanisms that drive the progression of breast cancer. Comput. Biol. Med. 2022, 145, 105508. [Google Scholar] [CrossRef]
- Dhirachaikulpanich, D.; Li, X.; Porter, L.F.; Paraoan, L. Integrated Microarray and RNAseq Transcriptomic Analysis of Retinal Pigment Epithelium/Choroid in Age-Related Macular Degeneration. Front. Cell Dev. Biol. 2020, 8, 808. [Google Scholar] [CrossRef]
- Nisar, M.; Paracha, R.Z.; Arshad, I.; Adil, S.; Zeb, S.; Hanif, R.; Rafiq, M.; Hussain, Z. Integrated Analysis of Microarray and RNA-Seq Data for the Identification of Hub Genes and Networks Involved in the Pancreatic Cancer. Front. Genet. 2021, 12, 663787. [Google Scholar] [CrossRef]
- Hozhabri, H.; Lashkari, A.; Razavi, S.M.; Mohammadian, A. Integration of gene expression data identifies key genes and path-ways in colorectal cancer. Med. Oncol. 2021, 38, 7. [Google Scholar] [CrossRef]
- Karimizadeh, E.; Sharifi-Zarchi, A.; Nikaein, H.; Salehi, S.; Salamatian, B.; Elmi, N.; Gharibdoost, F.; Mahmoudi, M. Analysis of gene expression profiles and protein-protein interaction networks in multiple tissues of systemic sclerosis. BMC Med. Genom. 2019, 12, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, D.; Galvez, J.M.; Herrera, L.J.; Roman, B.S.; Rojas, F.; Rojas, I. Integration of RNA-Seq data with heterogeneous microarray data for breast cancer profiling. BMC Bioinform. 2017, 18, 506. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Davis, M.S. Imports X: Package ‘GEOquery’. 2013. Available online: http://bioconductor.statistik.tu-dortmund.de/packages/2.12/bioc/manuals/GEOquery/man/GEOquery.pdf (accessed on 12 February 2022).
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Germain, D.R.; Mackey, J.R.; Godbout, R. Association of FABP5 expression with poor survival in triple-negative breast cancer: Implication for retinoic acid therapy. Am. J. Pathol. 2011, 178, 997–1008. [Google Scholar] [CrossRef]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M. Chronic oxidative stress promotes H2 AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef]
- Quigley, D.A.; Tahiri, A.; Luders, T.; Riis, M.H.; Balmain, A.; Borresen-Dale, A.L.; Bukholm, I.; Kristensen, V. Age, estrogen, and immune response in breast adenocarcinoma and adjacent normal tissue. Oncoimmunology 2017, 6, e1356142. [Google Scholar] [CrossRef]
- Lingle, W.; Erickson, B.; Zuley, M.; Jarosz, R.; Bonaccio, E.; Filippini, J.; Gruszauskas, N. Radiology data from the cancer genome atlas breast invasive carcinoma [TCGA-BRCA] collection. Cancer Imaging Arch. 2016, 10, K9. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Klaus, B.; Reisenauer, S. An end to end workflow for differential gene expression using Affymetrix microarrays. F1000Research 2016, 5, 1384. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, J.; Baranovskii, A.; Ronen, J.; Uyar, B.; Franke, V.; Akalin, A. Identifying tumor cells at the single-cell level using machine learning. Genome Biol. 2022, 23, 123. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Guo, X.; Yang, Y.-C.; Papcunik, D.; Heckman, C.; Hooke, J.; Shriver, C.D.; Liebman, M.N.; Hu, H. Detecting outlier microarray arrays by correlation and percentage of outliers spots. Cancer Inform. 2006, 2, 117693510600200017. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGA biolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-content normalization for RNA-Seq data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Hansen KD, Irizarry RA, Wu Z: Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 2012, 13, 204–216. [CrossRef] [PubMed]
- Zheng, W.; Chung, L.M.; Zhao, H. Bias detection and correction in RNA-Sequencing data. BMC Bioinform. 2011, 12, 290. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tomczak, A.; Mortensen, J.M.; Winnenburg, R.; Liu, C.; Alessi, D.T.; Swamy, V.; Vallania, F.; Lofgren, S.; Haynes, W.; Shah, N.H.; et al. Interpretation of biological experiments changes with evolution of the Gene Ontology and its annotations. Sci. Rep. 2018, 8, 5115. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic. Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Kolodner, R.D. An overview of Cdk1-controlled targets and processes. Cell Div. 2010, 5, 11. [Google Scholar] [CrossRef]
- Loukil, A.; Cheung, C.T.; Bendris, N.; Lemmers, B.; Peter, M.; Blanchard, J.M. Cyclin A2: At the crossroads of cell cycle and cell inva-sion. World J. Biol. Chem. 2015, 6, 346. [Google Scholar] [CrossRef]
- Androic, I.; Kramer, A.; Yan, R.; Rodel, F.; Gatje, R.; Kaufmann, M.; Strebhardt, K.; Yuan, J. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 2008, 8, 391. [Google Scholar] [CrossRef]
- Jezequel, P.; Campone, M.; Gouraud, W.; Guerin-Charbonnel, C.; Leux, C.; Ricolleau, G.; Campion, L. bc-GenExMiner: An easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res. Treat. 2012, 131, 765–775. [Google Scholar] [CrossRef]
- Jézéquel, P.; Frenel, J.-S.; Campion, L.; Guérin-Charbonnel, C.; Gouraud, W.; Ricolleau, G.; Campone, M. bc-GenExMiner 3.0: New mining module computes breast cancer gene expression correlation analyses. Database 2013, 2013, bas060. [Google Scholar] [CrossRef]
- Jezequel, P.; Gouraud, W.; Ben Azzouz, F.; Guerin-Charbonnel, C.; Juin, P.P.; Lasla, H.; Campone, M. bc-GenExMiner 4.5: New mining module computes breast cancer differential gene expression analyses. Database 2021, 2021, baab007. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Zhang, M.; Xu, D.Q.; Liu, Y.; Liu, T.; Liu, S.X.; Wang, P. Identification of hub genes and potential molecular mecha-nisms in gastric cancer by integrated bioinformatics analysis. PeerJ 2018, 6, e5180. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F. Hereditary breast cancer: The era of new susceptibility genes. Biomed. Res. Int. 2013, 2013, 747318. [Google Scholar] [CrossRef]
- Gage, M.; Wattendorf, D.; Henry, L. Translational advances regarding hereditary breast cancer syndromes. J. Surg. Oncol. 2012, 105, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Sonawane, M.D.; Song, K.S.; Kim, T. Biomarker detection technologies and future directions. Analyst 2016, 141, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Wu, M.; Yu, X.; Zhao, X.; Feng, Y.; Li, Y.; Lv, S. Identification of The Prognostic Genes for Early Basal-Like Breast Cancer with Weighted Gene Co-Expression Network Analysis. Medicine 2021, 101, e30581. [Google Scholar] [CrossRef]
- Toolabi, N.; Daliri, F.S.; Mokhlesi, A.; Talkhabi, M. Identification of key regulators associated with colon cancer prognosis and pathogenesis. J. Cell. Commun. Signal 2022, 16, 115–127. [Google Scholar] [CrossRef]
- Pan, Z.; Li, L.; Fang, Q.; Qian, Y.; Zhang, Y.; Zhu, J.; Ge, M.; Huang, P. Integrated Bioinformatics Analysis of Master Regulators in Anaplastic Thyroid Carcinoma. Biomed. Res. Int. 2019, 2019, 9734576. [Google Scholar] [CrossRef]
- Mo, X.C.; Zhang, Z.T.; Song, M.J.; Zhou, Z.Q.; Zeng, J.X.; Du, Y.F.; Sun, F.Z.; Yang, J.Y.; He, J.Y.; Huang, Y.; et al. Screening and identification of hub genes in bladder cancer by bioinformatics analysis and KIF11 is a potential prognostic biomarker. Oncol. Lett. 2021, 21, 205. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Mishra, A. Network analysis revealed aurora kinase dysregulation in five gynecological types of cancer. Oncol. Lett. 2018, 15, 1125–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Total Samples | Selected Samples | Platform | Country | Reference |
|---|---|---|---|---|---|
| GSE22820 | 186 samples Sample type: 10 Normal 176 Tumor | All | GPL6480 Agilent-014850 Whole Human Genome Microarray 4 × 44 K | Canada | Liu et al. [19] |
| GSE45827 | 155 samples Sample type: 11 Normal 14 Human Cell line 130 Tumor | 141 samples Sample type: 11 Normal 130 Tumor | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | France | Gruosso et al. [20] |
| GSE70905 | 137 samples Sample type: 47 Normal 43 Mammaplastic reduction 47 Tumor | 94 samples Sample type: 47 Normal 47 Tumor | GPL4133 Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F | USA | Quigley et al. [21] |
| TCGA-BRCA | 1208 samples Sample type: 113 Normal 1095 Tumor | All | Illumina HiSeq | Global | Pan-Cancer Atlas [16,22] |
| GO Term | p-Value | Odds Ratio | Combined Score |
|---|---|---|---|
| extracellular structure organization (GO:0043062) | 1.11266 × 10−12 | 5.849043 | 160.9906308 |
| external encapsulating structure organization (GO:0045229) | 1.25244 × 10−12 | 5.817629 | 159.4375212 |
| microtubule cytoskeleton organization involved in mitosis (GO:1902850) | 4.23453 × 10−12 | 7.754571 | 203.0747621 |
| extracellular matrix organization (GO:0030198) | 3.93235 × 10−11 | 4.510988 | 108.0796636 |
| positive regulation of cell cycle process (GO:0090068) | 1.64727 × 10−9 | 7.497769 | 151.6359712 |
| regulation of cell migration (GO:0030334) | 1.88723 × 10−9 | 3.546545 | 71.24355836 |
| mitotic spindle organization (GO:0007052) | 9.2914 × 10−9 | 5.424761 | 100.3264924 |
| endoderm formation (GO:0001706) | 2.56083 × 10−8 | 14.10305 | 246.5261963 |
| mitotic cytokinesis (GO:0000281) | 5.77801 × 10−8 | 10.6279 | 177.1311462 |
| positive regulation of protein kinase B signaling (GO:0051897) | 7.59979 × 10−8 | 4.961234 | 81.32732181 |
| GO Term | p-Value | Odds Ratio | Combined Score |
|---|---|---|---|
| protein homodimerization activity (GO:0042803) | 1.82 × 10−6 | 2.459593376 | 32.51094586 |
| platelet-derived growth factor binding (GO:0048407) | 5.68741 × 10−6 | 30.30218069 | 365.9672024 |
| microtubule motor activity (GO:0003777) | 1.7457 × 10−5 | 7.000721241 | 76.69830849 |
| microtubule binding (GO:0008017) | 1.89485 × 10−5 | 3.295333099 | 35.83274576 |
| tubulin binding (GO:0015631) | 3.43055 × 10−5 | 2.857481542 | 29.37549443 |
| motor activity (GO:0003774) | 6.71537 × 10−5 | 5.769550996 | 55.43688284 |
| actin binding (GO:0003779) | 9.57553 × 10−5 | 3.403527337 | 31.49526963 |
| receptor ligand activity (GO:0048018) | 9.98683 × 10−5 | 2.712680383 | 24.98828531 |
| cell-cell adhesion mediator activity (GO:0098632) | 1.19521 × 10−4 | 7.288930582 | 65.8337387 |
| metalloendopeptidase activity (GO:0004222) | 3.63166 × 10−4 | 4.501276991 | 35.65303972 |
| GO Term | p-Value | Odds Ratio | Combined Score |
|---|---|---|---|
| collagen-containing extracellular matrix (GO:0062023) | 4.04 × 10−12 | 4.231518 | 111.0119 |
| microtubule (GO:0005874) | 5.27 × 10−7 | 4.317358 | 62.40844 |
| spindle (GO:0005819) | 1.2 × 10−6 | 4.065692 | 55.43982 |
| polymeric cytoskeletal fibre (GO:0099513) | 1.93 × 10−6 | 3.489522 | 45.92285 |
| cell-substrate junction (GO:0030055) | 3.54 × 10−6 | 2.853014 | 35.81027 |
| spindle microtubule (GO:0005876) | 4.96 × 10−6 | 7.18054 | 87.70782 |
| focal adhesion (GO:0005925) | 7.4 × 10−6 | 2.792398 | 32.98852 |
| cell-cell junction (GO:0005911) | 1.54 × 10−4 | 2.779697 | 24.40338 |
| platelet alpha granule (GO:0031091) | 1.56 × 10−4 | 4.570755 | 40.05538 |
| actin-based cell projection (GO:0098858) | 3.98 × 10−4 | 4.44022 | 34.76533 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbashir, M.K.; Mohammed, M.; Mwambi, H.; Omolo, B. Identification of Hub Genes Associated with Breast Cancer Using Integrated Gene Expression Data with Protein-Protein Interaction Network. Appl. Sci. 2023, 13, 2403. https://doi.org/10.3390/app13042403
Elbashir MK, Mohammed M, Mwambi H, Omolo B. Identification of Hub Genes Associated with Breast Cancer Using Integrated Gene Expression Data with Protein-Protein Interaction Network. Applied Sciences. 2023; 13(4):2403. https://doi.org/10.3390/app13042403
Chicago/Turabian StyleElbashir, Murtada K., Mohanad Mohammed, Henry Mwambi, and Bernard Omolo. 2023. "Identification of Hub Genes Associated with Breast Cancer Using Integrated Gene Expression Data with Protein-Protein Interaction Network" Applied Sciences 13, no. 4: 2403. https://doi.org/10.3390/app13042403