Microglia: Agents of the CNS Pro-Inflammatory Response

 and
and
Abstract
:1. Introduction
2. Microglia Pro-Inflammatory Signaling
2.1. TLR4 Pro-Inflammatory Signaling
2.2. TLR4 Transcriptional Targets
2.3. TREM2 and Microglia Polarization States
2.4. Galectin-3 as a Hub Gene Associated to Neurodegeneration
3. Role of Caspases in Inflammatory Microglia Activation
3.1. Inflammasome and Pyroptosis
3.2. Necroptosis
3.3. Other Roles for Caspases During the Inflammatory Response
4. Novel Aspects of the Microglia Pro-Inflammatory Response
4.1. Microglia Communication via Extracellular Vesicles
4.2. Circadian Rhythm Regulates Microglia Pro-Inflammatory Response
5. Epigenetic Control of the Microglial Pro-Inflammatory Response
5.1. DNA Methylation during the Microglia Pro-Inflammation Response
5.2. Histone Modification and Microglia Activation
5.3. MicroRNAs and Pro-Inflammatory Microglia
6. Immunometabolism—The Link between Small Molecules and the Immune Response
6.1. Bioenergetics of Microglia: A Phenotypic Energy Switch
6.2. Amino Acid Metabolism and Immune Functions of mTOR
6.2.1. Arginine: A Fork in the Road Ahead
6.2.2. Tryptophan: The Two Fates of Kynurenine
6.2.3. Glutamine: The Alternative Energy Source
7. New Tools to Study Microglia Function
7.1. New Imaging Tools to Study Microglia
7.2. Human iPSCs as Source of Microglia
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ramón y Cajal, S. Contribución al Conocimiento de la Neuroglía del Cerebro Humano. Trab. Lab. Invest. Biol. Univ. Madrid 1913, XI, 215–315. [Google Scholar]
- Río-Hortega, P. Estudios sobre la neuroglía. La microglía y su transformación en células en bastoncito y cuerpos gránulo-adiposos. Trab. Lab. Invest. Biol. Univ. Madrid 1920, 18, 37–82. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. I. La microglía en estado normal. Bol. Soc. Esp. Biol. 1919, VIII, 67–82. [Google Scholar]
- Río-Hortega, P. El Tercer Elemento de los Centros Nerviosos. II. Intervención de la Microglía en los Procesos Patológicos (Células en Bastoncito y Cuerpos Gránuloadiposos). Bol. Soc. Esp. Biol. 1919, VIII, 91–103. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. III. Naturaleza probable de la microglía. Bol. Soc. Esp. Biol. 1919, VIII, 108–121. [Google Scholar]
- Río-Hortega, P. El “tercer elemento de los centros nerviosos”. IV. Poder fagocitario y movilidad de la microglía. Bol. Soc. Esp. Biol. 1919, VIII, 154–171. [Google Scholar]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Gomez-Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2014, 518, 547–551. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2016, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Aguilar, S.V.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; González, F.Z.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Venero, J.L.; Joseph, B.; Burguillos, M.A. Caspases orchestrate microglia instrumental functions. Prog. Neurobiol. 2018, 171, 50–71. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Paolicelli, R.C.; Kettenmann, H. Cien Años de Microglía: Milestones in a Century of Microglial Research. Trends Neurosci. 2019, 42, 778–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marı́n-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; Van Rooijen, N.; Mallat, M. Microglia Promote the Death of Developing Purkinje Cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.; Akiyama, H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci. Lett. 1988, 91, 259–264. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Van Horssen, J.; Singh, S.; Van Der Pol, S.M.A.; Kipp, M.; Lim, J.L.; Peferoen, L.; Gerritsen, W.H.; Kooi, E.-J.; Witte, M.E.; Geurts, J.J.; et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J. Neuroinflamm. 2012, 9, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickx, D.A.; Van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.-F.; Beach, T.G.; Tooyama, I. Microglial Phenotyping in Neurodegenerative Disease Brains: Identification of Reactive Microglia with an Antibody to Variant of CD105/Endoglin. Cells 2019, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.J.; Espinosa-Oliva, A.M.; Oliva-Martin, M.J.; Jiménez, A.C.; Venero, J.L.; De Pablos, R.M. Collateral Damage: Contribution of Peripheral Inflammation to Neurodegenerative Diseases. Curr. Top. Med. Chem. 2015, 15, 2193–2210. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2013, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Flowers, A.; Bell-Temin, H.; Jalloh, A.; Stevens, S.M.; Bickford, P.C. Proteomic analysis of aged microglia: Shifts in transcription, bioenergetics, and nutrient response. J. Neuroinflamm. 2017, 14, 96. [Google Scholar] [CrossRef]
- Galatro, T.; Holtman, I.R.; Lerario, A.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; Van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Ummenthum, K.M.D.; Peferoen, L.; Finardi, A.; Baker, D.; Pryce, G.; Mantovani, A.; Bsibsi, M.; Bottazzi, B.; Peferoen-Baert, R.; Van Der Valk, P.; et al. Pentraxin-3 is upregulated in the central nervous system during MS and EAE, but does not modulate experimental neurological disease. Eur. J. Immunol. 2015, 46, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Doni, A.; Peri, G.; Chieppa, M.; Allavena, P.; Pasqualini, F.; Vago, L.; Romani, L.; Garlanda, C.; Mantovani, A. Production of the soluble pattern recognition receptor PTX3 by myeloid, but not plasmacytoid, dendritic cells. Eur. J. Immunol. 2003, 33, 2886–2893. [Google Scholar] [CrossRef]
- Jeon, H.; Lee, S.; Lee, W.-H.; Suk, K. Analysis of glial secretome: The long pentraxin PTX3 modulates phagocytic activity of microglia. J. Neuroimmunol. 2010, 229, 63–72. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis is Sufficient to Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Rajbhandari, L.; Tegenge, M.A.; Shrestha, S.; Kumar, N.G.; Malik, A.; Mithal, A.; Hosmane, S.; Venkatesan, A. Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia 2014, 62, 1982–1991. [Google Scholar] [CrossRef]
- Koenigsknecht, J.; Landreth, G. Microglial Phagocytosis of Fibrillar β-Amyloid through a β1 Integrin-Dependent Mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicarlo, G.; Wilcock, D.; Henderson, D.; Gordon, M.; Morgan, D. Intrahippocampal LPS injections reduce AÎ2 load in APP+PS1 transgenic mice. Neurobiol. Aging 2001, 22, 1007–1012. [Google Scholar] [CrossRef]
- Herber, D.L.; Roth, L.M.; Wilson, D.; Wilson, N.; Mason, J.E.; Morgan, D.; Gordon, M. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Exp. Neurol. 2004, 190, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Raza, S.A.; Li, N.X.; Betarbet, R.; Dammer, E.B.; Duong, D.; Lah, J.J.; Seyfried, N.T.; Levey, A.I. Differential Phagocytic Properties of CD45low Microglia and CD45high Brain Mononuclear Phagocytes—Activation and Age-Related Effects. Front. Immunol. 2018, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.; Bateman, R.J. Decreased Clearance of CNS -Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.-Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [Green Version]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2016, 127, 624–633. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like receptor-mediated neuronal injury. Glia 2009, 58, 253–263. [Google Scholar] [CrossRef]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Akira, S. Toll-Like Receptors. Curr. Protoc. Immunol. 2015, 109, 14.12.1–14.12.10. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, O.; Braun, J.S.; Becker, D.; Halle, A.; Freyer, R.; Dagand, E.; Lehnardt, S.; Weber, J.R. TLR2 mediates neuroinflammation and neuronal damage. J. Immunol. 2007, 178, 6476–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Lee, H.-J.; Masliah, E.; Lee, S.J. Non-cell-autonomous Neurotoxicity of α-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 2016, 25, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar Amyloid-β Peptides Activate Microglia via TLR2: Implications for Alzheimer’s Disease1. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, S.; Cho, I.-H.; Lee, S.J. Toll-like receptors: Sensor molecules for detecting damage to the nervous system. Curr. Protein Pept. Sci. 2013, 14, 33–42. [Google Scholar] [CrossRef]
- De Oliveira, A.C.P.; Yousif, N.M.; Bhatia, H.S.; Hermanek, J.; Huell, M.; Fiebich, B.L. Poly(I:C) increases the expression of mPGES-1 and COX-2 in rat primary microglia. J. Neuroinflammation 2016, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA Is an Endogenous Ligand for Toll-like Receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bsibsi, M.; Bajramovic, J.; Vogt, M.H.J.; Van Duijvenvoorden, E.; Baghat, A.; Persoon-Deen, C.; Tielen, F.; Verbeek, R.; Huitinga, I.; Ryffel, B.; et al. The Microtubule Regulator Stathmin is an Endogenous Protein Agonist for TLR3. J. Immunol. 2010, 184, 6929–6937. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Liu, Y.; Hao, W.; Decker, Y.; Tomic, I.; Menger, M.D.; Liu, C.; Faßbender, K. Stimulation of TLR4 Attenuates Alzheimer’s Disease–Related Symptoms and Pathology in Tau-Transgenic Mice. J. Immunol. 2016, 197, 3281–3292. [Google Scholar] [CrossRef]
- Venezia, S.; Refolo, V.; Polissidis, A.; Stefanis, L.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal α-synucleinopathy. Mol. Neurodegener. 2017, 12, 52. [Google Scholar] [CrossRef]
- Song, M.; Jin, J.; Lim, J.-E.; Kou, J.; Pattanayak, A.; Rehman, J.; Kim, H.-D.; Tahara, K.; LaLonde, R.; Fukuchi, K.-I. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, L.; Irschick, R.B.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2012, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Yan, W.-F.; Zhang, Z.; Ma, K.-L.; Peng, S.-Y.; Cao, Y.-L.; Yuan, Y.-H.; Chen, N.-H. Nurr1: A vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology 2019, 144, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Weng, R.; Chen, Z.; Wang, R.; Zou, J.; Liu, X.; Liao, J.; Wang, Y.; Xia, Y.; Wang, Q. TLR4 Signaling in MPP+-Induced Activation of BV-2 Cells. Neural Plast. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstens, E.; Akiyama, T. Itch: Mechanisms and Treatment; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2014. [Google Scholar]
- Rosenberger, K.; Derkow, K.; Dembny, P.; Krüger, C.; Schott, E.; Lehnardt, S. The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J. Neuroinflamm. 2014, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; David, K.; et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Lehmann, S.M.; Rosenberger, K.; Krüger, C.; Habbel, P.; Derkow, K.; Kaul, D.; Rybak, A.; Brandt, C.; Schott, E.; Wulczyn, F.G.; et al. Extracellularly Delivered Single-Stranded Viral RNA Causes Neurodegeneration Dependent on TLR7. J. Immunol. 2012, 189, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Tauber, S.C.; Ebert, S.; Weishaupt, J.H.; Reich, A.; Nau, R.; Gerber, J. Stimulation of Toll-Like Receptor 9 by Chronic Intraventricular Unmethylated Cytosine-Guanine DNA Infusion Causes Neuroinflammation and Impaired Spatial Memory. J. Neuropathol. Exp. Neurol. 2009, 68, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.; Gerber, J.; Bader, S.; Mühlhauser, F.; Brechtel, K.; Mitchell, T.J.; Nau, R. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J. Neuroimmunol. 2005, 159, 87–96. [Google Scholar] [CrossRef]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J. Neurosci. 2009, 29, 1846–1854. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Murao, N.; Katano, Y.; Juliandi, B.; Kohyama, J.; Akira, S.; Kawai, T.; Nakashima, K. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat. Commun. 2015, 6, 6514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.M.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, H.T.; Kul, O.; Karakuş, E.; Terzi, O.; Canpolat, S.; Anteplioğlu, T. Astrocytes, microglia/macrophages, and neurons expressing Toll-like receptor 11 contribute to innate immunity against encephalitic Toxoplasma gondii infection. Neuroscience 2014, 269, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Kolter, J.; Feuerstein, R.; Spoeri, E.; Gharun, K.; Elling, R.; Trieu-Cuot, P.; Goldmann, T.; Waskow, C.; Chen, Z.J.; Kirschning, C.J.; et al. Streptococci Engage TLR13 on Myeloid Cells in a Site-Specific Fashion. J. Immunol. 2016, 196, 2733–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Moriguchi, T.; Kuroyanagi, N.; Yamaguchi, K.; Gotoh, Y.; Irie, K.; Kano, T.; Shirakabe, K.; Muro, Y.; Shibuya, H.; Matsumoto, K.; et al. A Novel Kinase Cascade Mediated by Mitogen-activated Protein Kinase Kinase 6 and MKK3. J. Biol. Chem. 1996, 271, 13675–13679. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, K.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Matsuda, S.; Moriguchi, T.; Gotoh, Y.; Matsumoto, K.; Nishida, E. TAK1 Mediates the Ceramide Signaling to Stress-activated Protein Kinase/c-Jun N-terminal Kinase. J. Biol. Chem. 1997, 272, 8141–8144. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.-I.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Offermann, M.K. Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF is Dependent on its Receptor Interacting Protein Homotypic Interaction Motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.; Jin, W.; Sun, S.-C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medvedev, A.E.; Murphy, M.; Zhou, H.; Li, X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol. Rev. 2015, 266, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.-H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Jiang, Z. The essential adaptors of innate immune signaling. Protein Cell 2012, 4, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Sedger, L.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [Green Version]
- Song, H.Y.; Régnier, C.H.; Kirschning, C.J.; Goeddel, D.V.; Rothe, M. Tumor necrosis factor (TNF)-mediated kinase cascades: Bifurcation of Nuclear Factor- B and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl. Acad. Sci. USA 1997, 94, 9792–9796. [Google Scholar] [CrossRef] [Green Version]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2016, 174, 1733–1749. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2017, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.-J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef]
- Terazawa, R.; Akimoto, N.; Kato, T.; Itoh, T.; Fujita, Y.; Hamada, N.; Deguchi, T.; Iinuma, M.; Noda, M.; Nozawa, Y.; et al. A kavalactone derivative inhibits lipopolysaccharide-stimulated iNOS induction and NO production through activation of Nrf2 signaling in BV2 microglial cells. Pharmacol. Res. 2013, 71, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of Inducible Nitric Oxide Synthase Gene in Glial Cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflammation 2005, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.-Y.; Shelhamer, J.H. Toll-like Receptor 4 Signaling Regulates Cytosolic Phospholipase A2Activation and Lipid Generation in Lipopolysaccharide-stimulated Macrophages. J. Biol. Chem. 2005, 280, 38969–38975. [Google Scholar] [CrossRef] [Green Version]
- Hein, A.M.; O’Banion, M.K. Neuroinflammation and Memory: The Role of Prostaglandins. Mol. Neurobiol. 2009, 40, 15–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Ran, Y.; Qie, S.; Gong, W.; Gao, F.; Ding, Z.; Xi, J. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yuan, M.; Wang, S.; Zhang, L.; Zhang, R.; Zou, X.; Wang, X.; Chen, D.; Wu, Z. STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1. Int. J. Mol. Sci. 2019, 20, 4870. [Google Scholar] [CrossRef] [Green Version]
- Porro, C.; Cianciulli, A.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Curcumin Regulates Anti-Inflammatory Responses by JAK/STAT/SOCS Signaling Pathway in BV-2 Microglial Cells. Biology 2019, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Li, S.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Zhang, A.; Jia, Z.; Li, M. COX-2 contributes to LPS-induced Stat3 activation and IL-6 production in microglial cells. Am. J. Transl. Res. 2018, 10, 966–974. [Google Scholar]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Jónsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, T.; Von Saucken, V.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2)*. J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.C.; Devaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E*. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [Green Version]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Ferrer, I.J.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835.e7. [Google Scholar] [CrossRef]
- Walker, D.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.-F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.-H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nyström, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Kumar, N.T.; Liestøl, K.; Løberg, E.M.; Reims, H.; Mæhlen, J. Apolipoprotein E allelotype is associated with neuropathological findings in Alzheimer’s disease. Virchows Arch. 2015, 467, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ruberu, N.N.; Harada, M.; Arai, T.; Sawabe, M.; Nukina, N.; Murayama, S. In situ detection of apolipoprotein E ε4 in archival human brain. NeuroReport 2004, 15, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.; Hirsch, A.M.; Broihier, M.L.; Miller, C.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2016, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Initiative, A.D.N.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Minami, S.S.; Min, S.-W.; Krabbe, G.; Wang, C.; Zhou, Y.; Asgarov, R.; Li, Y.; Martens, L.H.; Elia, L.P.; Ward, M.E.; et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014, 20, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Götzl, J.K.; Brendel, M.; Werner, G.; Parhizkar, S.; Monasor, L.S.; Kleinberger, G.; Colombo, A.; Deussing, M.; Wagner, M.; Winkelmann, J.; et al. Opposite microglial activation stages upon loss of PGRN or TREM 2 result in reduced cerebral glucose metabolism. EMBO Mol. Med. 2019, 11, e9711. [Google Scholar] [CrossRef]
- Hosokawa, M.; Arai, T.; Masuda-Suzukake, M.; Kondo, H.; Matsuwaki, T.; Nishihara, M.; Hasegawa, M.; Akiyama, H. Progranulin Reduction is Associated with Increased Tau Phosphorylation in P301L Tau Transgenic Mice. J. Neuropathol. Exp. Neurol. 2015, 74, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Monasor, L.S.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 2020, 9. [Google Scholar] [CrossRef]
- Cerliani, J.P.; Blidner, A.G.; Toscano, M.A.; Croci, D.O.; Rabinovich, G.A. Translating the ‘Sugar Code’ into Immune and Vascular Signaling Programs. Trends Biochem. Sci. 2017, 42, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Leffler, H.; Carlsson, S.; Hedlund, M.; Qian, Y.; Poirier, F. Introduction to galectins. Glycoconj. J. 2002, 19, 433–440. [Google Scholar] [CrossRef]
- Yip, P.K.; Jiménez, A.C.; King, P.; Vilalta, A.; Nomura, K.; Chau, C.C.; Egerton, A.M.S.; Liu, Z.-H.; Shetty, A.J.; Tremoleda, J.L.; et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 2017, 7, 41689. [Google Scholar] [CrossRef] [Green Version]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; García-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Lalancette-Hebert, M.; Swarup, V.; Beaulieu, J.-M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 Is Required for Resident Microglia Activation and Proliferation in Response to Ischemic Injury. J. Neurosci. 2012, 32, 10383–10395. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Allendorf, D.H. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boza-Serrano, A.; Reyes, J.F.; Rey, N.L.; Leffler, H.; Bousset, L.; Nilsson, U.J.; Brundin, P.; Venero, J.L.; Burguillos, M.A.; Deierborg, T. The role of Galectin-3 in α-synuclein-induced microglial activation. Acta Neuropathol. Commun. 2014, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Mostacada, K.; De Oliveira, F.L.; Villa-Verde, D.M.; Martinez, A.M.B. Lack of galectin-3 improves the functional outcome and tissue sparing by modulating inflammatory response after a compressive spinal cord injury. Exp. Neurol. 2015, 271, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Siew, J.J.; Chen, H.-M.; Chen, H.-Y.; Chen, H.-L.; Chen, C.-M.; Soong, B.-W.; Wu, Y.-R.; Chang, C.-P.; Chan, Y.-C.; Lin, C.-H.; et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 2019, 10, 3473–3518. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Henry, C.M.; Cullen, S.P. A Perspective on Mammalian Caspases as Positive and Negative Regulators of Inflammation. Mol. Cell 2012, 46, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Pop, C.; Salvesen, G.S. Human Caspases: Activation, Specificity, and Regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [Green Version]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2014, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.L.; Stutz, A.; Alnemri, E.S.; Macdonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Ozkurede, U.; Kim, Y.-G.; Arindam, C.; Gale, M.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 2014, 193, 4214–4222. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.; Mitoma, H.; Hanabuchi, S.; Bao, M.; Weng, L.; Sugimoto, N.; Liu, Y.; Zhang, Z.; Zhong, J.; Sun, B.; et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 16059–16064. [Google Scholar] [CrossRef] [Green Version]
- Brož, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Hutchinson, M.R.; Bilbo, S.D. Early-Life Experience Decreases Drug-Induced Reinstatement of Morphine CPP in Adulthood via Microglial-Specific Epigenetic Programming of Anti-Inflammatory IL-10 Expression. J. Neurosci. 2011, 31, 17835–17847. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Investig. 2018, 128, 5399–5412. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.L.; Haley, B.; Roose-Girma, M.; Phung, Q.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2009, 17, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2018, 20, 19–33. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.M.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitination in Signaling to and Activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, F.; Wang, X. TNF-α Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; De Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Gonçalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; Macfarlane, M.; Häcker, G.; Leverkus, M. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; Macfarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Wachter, T.; Sprick, M.R.; Hausmann, D.; Kerstan, A.; McPherson, K.; Stassi, G.; Bröcker, E.-B.; Walczak, H.; Leverkus, M. cFLIPLInhibits Tumor Necrosis Factor-related Apoptosis-inducing Ligand-mediated NF-κB Activation at the Death-inducing Signaling Complex in Human Keratinocytes. J. Biol. Chem. 2004, 279, 52824–52834. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The Role of c-FLIP in Modulation of CD95-induced Apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.E.; Spangenberg, E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, J.J.; Cogswell, P.; Flavell, R.A.; Baldwin, J.A.S.; Davis, R.J. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-S.; Morgan, M.J.; Choksi, S.; Liu, Z.-G. TNF-Induced Activation of the Nox1 NADPH Oxidase and its Role in the Induction of Necrotic Cell Death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef]
- Zhang, D.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator that Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.-Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nature 2018, 20, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Vilalta, A.; Tolkovsky, A.M.; Brown, G.C. Caspase Inhibitors Protect Neurons by Enabling Selective Necroptosis of Inflamed Microglia. J. Biol. Chem. 2013, 288, 9145–9152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2017, 25, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; García-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Kavanagh, E.; Rodhe, J.; Burguillos, M.A.; Venero, J.L.; Joseph, B. Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death Dis. 2014, 5, e1565. [Google Scholar] [CrossRef] [Green Version]
- Berthelet, J.; Dubrez, L. Regulation of Apoptosis by Inhibitors of Apoptosis (IAPs). Cells 2013, 2, 163–187. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, E.; Burguillos, M.A.; Jiménez, A.C.; Oliva-Martin, M.J.; Santiago, M.; Rodhe, J.; Joseph, B.; Venero, J.L. Deletion of caspase-8 in mouse myeloid cells blocks microglia pro-inflammatory activation and confers protection in MPTP neurodegeneration model. Aging 2015, 7, 673–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.K.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1β and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [Green Version]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; Van Der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B.H. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.-L.; Gentle, I.E.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Groß, O.; Ma, S.; Guarda, G.; et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Hopkins, L.J.; Nugent, E.; Cox, S.; Glück, I.M.; Tourlomousis, P.; Wright, J.A.; Cicuta, P.; Monie, T.P.; Bryant, C.E. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 2014, 111, 7403–7408. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.S.; Walle, L.V.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Vince, J.E. Ambiguities in NLRP3 inflammasome regulation: Is there a role for mitochondria? Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Bartok, E.; Gaidt, M.M.; Bock, F.J.; Herrmann, J.; Seeger, J.M.; Brož, P.; Beckmann, R.; Kashkar, H.; Tait, S.W.; et al. BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1β Maturation in Macrophages. Cell Rep. 2018, 25, 2354–2368.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.; McArthur, K.; Metcalf, N.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; Van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmuganathan, M.; Vughs, J.; Noseda, M.; Emanueli, C. Exosomes: Basic Biology and Technological Advancements Suggesting Their Potential as Ischemic Heart Disease Therapeutics. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated Exosome Secretion Promotes Clearance of Amyloid-β by Microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef] [Green Version]
- Fitzner, D.; Schnaars, M.; Van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.-K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes Derived from Epstein-Barr Virus-Infected Cells are Internalized via Caveola-Dependent Endocytosis and Promote Phenotypic Modulation in Target Cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012, 31, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonski, K.A.; Gaudet, A.; Amici, S.A.; Popovich, P.G.; Guerau-De-Arellano, M. Control of the Inflammatory Macrophage Transcriptional Signature by miR-155. PLoS ONE 2016, 11, e0159724. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Stoica, B.A.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A.I. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflammation 2017, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.-I.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with hSOD1-G93A Are Enriched in miR-124 and Drive Alterations in Microglia Phenotype. Front. Mol. Neurosci. 2017, 11, 273. [Google Scholar] [CrossRef] [Green Version]
- Harrison, E.; Hochfelder, C.G.; Lamberty, B.G.; Meays, B.M.; Morsey, B.M.; Kelso, M.L.; Fox, H.S.; Yelamanchili, S.V. Traumatic brain injury increases levels of miR-21 in extracellular vesicles: Implications for neuroinflammation. FEBS Open Bio 2016, 6, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Tapia, R.J.; Chavarría, A.; Navarro, L.; Anahí, C.; Luz, N. Differences in Diurnal Variation of Immune Responses in Microglia and Macrophages: Review and Perspectives. Cell. Mol. Neurobiol. 2019, 40, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.; Weil, Z.; Nelson, R.J. Mice exposed to dim light at night exaggerate inflammatory responses to lipopolysaccharide. Brain Behav. Immun. 2013, 34, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.K.; Frank, M.G.; Kitt, M.M.; Barrientos, R.M.; Watkins, L.R.; Maier, S.F. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav. Immun. 2014, 45, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonken, L.K.; Kitt, M.M.; Gaudet, A.D.; Barrientos, R.M.; Watkins, L.R.; Maier, S.F. Diminished circadian rhythms in hippocampal microglia may contribute to age-related neuroinflammatory sensitization. Neurobiol. Aging 2016, 47, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Griffin, P.; Dimitry, J.M.; Sheehan, P.W.; Lananna, B.V.; Guo, C.; Robinette, M.L.; Hayes, M.E.; Cedeño, M.R.; Nadarajah, C.J.; Ezerskiy, L.A.; et al. Circadian clock protein Rev-erbα regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 5102–5107. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, D.; Griffin, P.; Sheehan, P.W.; Kim, D.; Musiek, E.; Yoon, S.-Y. Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell 2019, 19, e13078. [Google Scholar] [CrossRef] [Green Version]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular Signals of Epigenetic States. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Jimenez, A.; Deniz, Ö.; Niklison-Chirou, M.V.; Ruiz, R.; Bezerra-Salomão, K.; Stratoulias, V.; Amouroux, R.; Yip, P.K.; Vilalta, A.; Cheray, M.; et al. TET2 Regulates the Neuroinflammatory Response in Microglia. Cell Rep. 2019, 29, 697–713.e8. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2011, 13, 7–13. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Zhang, Z.; Fauser, U.; Schluesener, H.J. Global hypomethylation defines a sub-population of reactive microglia/macrophages in experimental traumatic brain injury. Neurosci. Lett. 2007, 429, 1–6. [Google Scholar] [CrossRef]
- Matt, S.; Lawson, M.A.; Johnson, R. Aging and peripheral lipopolysaccharide can modulate epigenetic regulators and decrease IL-1β promoter DNA methylation in microglia. Neurobiol. Aging 2016, 47, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-H.; Chen, J.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.-S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Lio, C.J.; Samaniego-Castruita, D.; Li, X.; Rao, A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat. Commun. 2019, 10, 2011. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Oliva, A.M.; Burguillos, M.A. TET2, an “ambiguous” player in inflammation. Neural Regen. Res. 2020, 15, 1481–1482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Cochran, J.N.; Geier, E.G.; Bonham, L.W.; Newberry, J.S.; Amaral, M.D.; Thompson, M.L.; Lasseigne, B.N.; Karydas, A.M.; Roberson, E.D.; Cooper, G.M.; et al. Non-coding and Loss-of-Function Coding Variants in TET2 are Associated with Multiple Neurodegenerative Diseases. Am. J. Hum. Genet. 2020, 106, 632–645. [Google Scholar] [CrossRef]
- Khorasanizadeh, S. The Nucleosome. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Jiang, X. Histone Deacetylase Inhibitors in Programmed Cell Death and Cancer Therapy. Cell Cycle 2005, 4, 549–551. [Google Scholar] [CrossRef]
- Stewart-Morgan, K.R.; Petryk, N.; Groth, A. Chromatin replication and epigenetic cell memory. Nature 2020, 22, 361–371. [Google Scholar] [CrossRef]
- Blanchard, F.; Kinzie, E.; Wang, Y.; Duplomb, L.; Godard, A.; Held, W.A.; Asch, B.B.; Tamiaki, H. FR901228, an inhibitor of histone deacetylases, increases the cellular responsiveness to IL-6 type cytokines by enhancing the expression of receptor proteins. Oncogene 2002, 21, 6264–6277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Lim, S.; Caramori, G.; Cosio, B.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Lipska, K.; Gumieniczek, A.; Filip, A.A. Anticonvulsant valproic acid and other short-chain fatty acids as novel anticancer therapeutics: Possibilities and challenges. Acta Pharm. 2020, 70, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, V.; Brouwer, N.; Hanisch, U.-K.; Regen, T.; Eggen, B.J.; Boddeke, H.W.G.M. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J. Neurosci. Res. 2013, 91, 1133–1142. [Google Scholar] [CrossRef]
- Suh, H.-S.; Choi, S.; Khattar, P.; Choi, N.; Lee, S.C. Histone Deacetylase Inhibitors Suppress the Expression of Inflammatory and Innate Immune Response Genes in Human Microglia and Astrocytes. J. Neuroimmune Pharmacol. 2010, 5, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, B.S.; Grigg, R.; Wood, I.C. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J. Neurochem. 2017, 143, 214–224. [Google Scholar] [CrossRef]
- Fleiss, B.; Nilsson, M.K.; Blomgren, K.; Mallard, C. Neuroprotection by the histone deacetylase inhibitor trichostatin A in a model of lipopolysaccharide-sensitised neonatal hypoxic-ischaemic brain injury. J. Neuroinflamm. 2012, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia during Ischemic Stroke. Mol. Neurobiol. 2016, 54, 6391–6411. [Google Scholar] [CrossRef]
- Datta, M.; Staszewski, O.; Raschi, E.; Frosch, M.; Hagemeyer, N.; Tay, T.L.; Blank, T.; Kreutzfeldt, M.; Merkler, D.; Ziegler-Waldkirch, S.; et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity 2018, 48, 514–529.e6. [Google Scholar] [CrossRef]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saidi, D.; Cheray, M.; Osman, A.M.; Stratoulias, V.; Lindberg, O.R.; Shen, X.; Blomgren, K.; Joseph, B. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. OncoImmunology 2017, 7, e1382790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2013, 21, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arifuzzaman, S.; Das, A.; Kim, S.H.; Yoon, T.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. Selective inhibition of EZH2 by a small molecule inhibitor regulates microglial gene expression essential for inflammation. Biochem. Pharmacol. 2017, 137, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef]
- Masuda, T.; Iwamoto, S.; Mikuriya, S.; Tozaki-Saitoh, H.; Tamura, T.; Tsuda, M.; Inoue, K. Transcription factor IRF1 is responsible for IRF8-mediated IL-1β expression in reactive microglia. J. Pharmacol. Sci. 2015, 128, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Christoforidou, E.; Joilin, G.; Hafezparast, M. Potential of activated microglia as a source of dysregulated extracellular microRNAs contributing to neurodegeneration in amyotrophic lateral sclerosis. J. Neuroinflamm. 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonté, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Guedes, J.R.; De Almeida, L.P.; De Lima, M.C.P. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology 2011, 135, 73–88. [Google Scholar] [CrossRef]
- Koval, E.D.; Shaner, C.; Zhang, P.; Du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2014, 77, 75–99. [Google Scholar] [CrossRef]
- Yip, P.K.; Bowes, A.L.; Hall, J.C.E.; Burguillos, M.A.; Ip, T.H.R.; Baskerville, T.; Liu, Z.-H.; Mohamed, M.A.E.K.; Getachew, F.; Lindsay, A.D.; et al. Docosahexaenoic acid reduces microglia phagocytic activity via miR-124 and induces neuroprotection in rodent models of spinal cord contusion injury. Hum. Mol. Genet. 2019, 28, 2427–2448. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Song, J.; Oh, Y.; Lee, W.T. MicroRNA-Let-7a regulates the function of microglia in inflammation. Mol. Cell. Neurosci. 2015, 68, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, X.; Chen, S.; Liu, H.; Wang, Y.; Xu, X.; Cheng, J.; Jia, J.; Zhen, X. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav. Immun. 2015, 49, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zeng, Y.; Qian, Y.; Dong, J.; Zhang, Z.; Zhang, J. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain Res. 2018, 1685, 91–104. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J.C. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Luan, H.H.; Medzhitov, R. An evolutionary perspective on immunometabolism. Science 2019, 363, eaar3932. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Aldana, B.I.; Nissen, J.D.; Tanila, H.; Waagepetersen, H.S. Alterations in Cerebral Cortical Glucose and Glutamine Metabolism Precedes Amyloid Plaques in the APPswe/PSEN1dE9 Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2016, 42, 1589–1598. [Google Scholar] [CrossRef]
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Chaudhuri, K.R.; Walker, Z.; Turkheimer, F.; Brooks, D.J. Microglia, Amyloid, and Glucose Metabolism in Parkinson’s Disease with and without Dementia. Neuropsychopharmacology 2013, 38, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013, 587, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauro, C.; Chece, G.; Monaco, L.; Antonangeli, F.; Peruzzi, G.; Rinaldo, S.; Paone, A.; Cutruzzolà, F.; Limatola, C. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front. Cell. Neurosci. 2019, 13, 414. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2017, 301, 120–132. [Google Scholar] [CrossRef]
- Silva, A.B.D.P.E.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflamm. 2019, 16, 152. [Google Scholar] [CrossRef] [Green Version]
- Chausse, B.; Kakimoto, P.A.; Caldeira-Da-Silva, C.C.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; Da Silva, R.P.; Miyamoto, S.; Kowaltowski, A.J. Distinct metabolic patterns during microglial remodeling by oleate and palmitate. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Cordes, T.; Lucas, A.; Divakaruni, A.S.; Murphy, A.N.; Cabrales, P.; Metallo, C.M. Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Mol. Metab. 2020, 32, 122–135. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannahill, G.M.; Curtis, A.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Strelko, C.L.; Lu, W.; Dufort, F.J.; Seyfried, T.N.; Chiles, T.C.; Rabinowitz, J.D.; Roberts, M.F. Itaconic Acid Is a Mammalian Metabolite Induced during Macrophage Activation. J. Am. Chem. Soc. 2011, 133, 16386–16389. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Iacobazzi, V.; Menga, A.; Avantaggiati, M.L.; Palmieri, F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim. Biophys. Acta Bioenerg. 2014, 1839, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein is an Adaptor that Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.-J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J. Neuroinflammation 2020, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, I.N.; Shperdheja, J.; Baybis, M.; Ferguson, T.; Crino, P.B. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol. Dis. 2016, 85, 144–154. [Google Scholar] [CrossRef]
- Xie, L.; Sun, F.; Wang, J.; Mao, X.; Xie, L.; Yang, S.-H.; Su, D.-M.; Simpkins, J.W.; Greenberg, D.A.; Jin, K. mTOR Signaling Inhibition Modulates Macrophage/Microglia-Mediated Neuroinflammation and Secondary Injury via Regulatory T Cells after Focal Ischemia. J. Immunol. 2014, 192, 6009–6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nature 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.-F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Lei, Q.; Tan, J.; Yi, S.; Wu, N.; Wang, Y.; Wu, H. Mitochonic acid 5 activates the MAPK–ERK–yap signaling pathways to protect mouse microglial BV-2 cells against TNFα-induced apoptosis via increased Bnip3-related mitophagy. Cell. Mol. Biol. Lett. 2018, 23, 14. [Google Scholar] [CrossRef]
- Ye, J.; Jiang, Z.; Chen, X.; Liu, M.; Li, J.; Liu, N. The role of autophagy in pro-inflammatory responses of microglia activation via mitochondrial reactive oxygen speciesin vitro. J. Neurochem. 2017, 142, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Bernier, L.-P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Kawahara, K.; Gotoh, T.; Oyadomari, S.; Kajizono, M.; Kuniyasu, A.; Ohsawa, K.; Imai, Y.; Kohsaka, S.; Nakayama, H.; Mori, M. Co-induction of argininosuccinate synthetase, cationic amino acid transporter-2, and nitric oxide synthase in activated murine microglial cells. Mol. Brain Res. 2001, 90, 165–173. [Google Scholar] [CrossRef]
- Sawano, T.; Tsuchihashi, R.; Watanabe, F.; Niimi, K.; Yamaguchi, W.; Yamaguchi, N.; Furuyama, T.; Tanaka, H.; Matsuyama, T.; Inagaki, S. Changes in L-arginine metabolism by Sema4D deficiency induce promotion of microglial proliferation in ischemic cortex. Neuroscience 2019, 406, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Chang, C.-C.; Chang, T.-S.; Li, H.-H.; Hung, H.-C.; Liu, G.-Y.; Lin, C.-L. Aβ stimulates microglial activation through antizyme-dependent downregulation of ornithine decarboxylase. J. Cell. Physiol. 2018, 234, 9733–9745. [Google Scholar] [CrossRef] [PubMed]
- Bagasra, O.; Michaels, F.H.; Zheng, Y.M.; Bobroski, L.E.; Spitsin, S.V.; Fu, Z.F.; Tawadros, R.; Koprowski, H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 12041–12045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Thomas, T.J. Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001, 58, 244–258. [Google Scholar] [CrossRef]
- Klimaszewska-Łata, J.; Gul-Hinc, S.; Bielarczyk, H.; Ronowska, A.; Zyśk, M.; Grużewska, K.; Pawełczyk, T.; Szutowicz, A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J. Neurochem. 2015, 133, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Moffett, J.R.; Arun, P.; Puthillathu, N.; Vengilote, R.; Ives, J.A.; Badawy, A.A.-B.; Namboodiri, A.M. Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD+ Synthesis During Inflammation and Infection. Front. Immunol. 2020, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.L.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2004, 49, 15–23. [Google Scholar] [CrossRef]
- Tao, X.; Yan, M.; Wang, L.; Zhou, Y.; Wang, Z.; Xia, T.; Liu, X.; Pan, R.; Chang, Q. Homeostasis Imbalance of Microglia and Astrocytes Leads to Alteration in the Metabolites of the Kynurenine Pathway in LPS-Induced Depressive-Like Mice. Int. J. Mol. Sci. 2020, 21, 1460. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Borza, I.; Kolok, S.; Galgóczy, K.; Gere, A.; Horváth, C.; Farkas, S.; Greiner, I.; Domány, G. Kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorganic Med. Chem. Lett. 2007, 17, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Chretien, F.; Vallat-Decouvelaere, A.; Bossuet, C.; Rimaniol, A.-C.; Le Grand, R.; Le Pavec, G.; Creminon, C.; Dormont, D.; Gray, F.; Gras, G. Expression of excitatory amino acid transporter-2 (EAAT-2) and glutamine synthetase (GS) in brain macrophages and microglia of SIVmac251-infected macaques. Neuropathol. Appl. Neurobiol. 2002, 28, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanamatsu, T.; Takezawa, Y.; Kohsaka, S. Up-regulation of glutamine synthesis in microglia activated with endotoxin. Neurosci. Lett. 2015, 591, 99–104. [Google Scholar] [CrossRef]
- Nagy, A.M.; Fekete, R.; Horvath, G.; Koncsos, G.; Kriston, C.; Sebestyén, A.; Giricz, Z.; Környei, Z.; Madarász, E.; Tretter, L. Versatility of microglial bioenergetic machinery under starving conditions. Biochim. et Biophys. Acta Bioenerg. 2018, 1859, 201–214. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxid. Redox Signal. 2017, 26, 351–363. [Google Scholar] [CrossRef]
- Niklison-Chirou, M.V.; Erngren, I.; Engskog, M.; Haglöf, J.; Picard, D.; Remke, M.; McPolin, P.H.R.; Selby, M.; Williamson, D.; Clifford, S.C.; et al. TAp73 is a marker of glutamine addiction in medulloblastoma. Genes Dev. 2017, 31, 1738–1753. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Du, X.-F.; Liu, C.-S.; Wen, Z.; Du, J.-L. Reciprocal Regulation between Resting Microglial Dynamics and Neuronal Activity In Vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2019, 367, 528–537. [Google Scholar] [CrossRef]
- Sharma, K.; Wu, L.-J.; Eyo, U.B. Calming Neurons with a Microglial Touch. Trends Neurosci. 2020, 43, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Masgrau, R.; Guaza, C.; Ransohoff, R.M.; Galea, E. Should We Stop Saying ‘Glia’ and ‘Neuroinflammation’? Trends Mol. Med. 2017, 23, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Is ‘friendly fire’ in the brain provoking Alzheimer’s disease? Nature 2018, 556, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef] [Green Version]
- The National Academies of Sciences, Engineering and Medicine. Biomarkers of Neuroinflammation—Proceedings of a Workshop; The National Academies Press: Washington, DC, USA, 2018. [Google Scholar]
- Albrecht, D.S.; Granziera, C.; Hooker, J.M.; Loggia, M.L. In Vivo Imaging of Human Neuroinflammation. ACS Chem. Neurosci. 2016, 7, 470–483. [Google Scholar] [CrossRef] [Green Version]
- Cosenza-Nashat, M.; Zhao, M.-L.; Suh, H.-S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [Green Version]
- Abourbeh, G.; Thézé, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dollé, F.; Tavitian, B.; Boisgard, R. Imaging Microglial/Macrophage Activation in Spinal Cords of Experimental Autoimmune Encephalomyelitis Rats by Positron Emission Tomography using the Mitochondrial 18 kDa Translocator Protein Radioligand [18F]DPA-714. J. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef] [Green Version]
- Amhaoul, H.; Hamaide, J.; Bertoglio, D.; Reichel, S.N.; Verhaeghe, J.; Geerts, E.; Van Dam, D.; De Deyn, P.P.; Kumar-Singh, S.; Katsifis, A.; et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol. Dis. 2015, 82, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.; Beck, R.; Lindner, S.; Gildehaus, F.-J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, I.; Ohsiek, A.; Al-Momani, E.; Albert-Weissenberger, C.; Stetter, C.; Mencl, S.; Buck, A.K.; Kleinschnitz, C.; Samnick, S.; Sirén, A.-L. Combined [18F]DPA-714 micro-positron emission tomography and autoradiography imaging of microglia activation after closed head injury in mice. J. Neuroinflamm. 2016, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, A.; Boisgard, R.; Thézé, B.; Van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dollé, F.; Tavitian, B. Evaluation of the PBR/TSPO Radioligand [18F]DPA-714 in a Rat Model of Focal Cerebral Ischemia. Br. J. Pharmacol. 2009, 30, 230–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa Translocator Protein (TSPO) Polymorphism Explains Differences in Binding Affinity of the PET Radioligand PBR28. Br. J. Pharmacol. 2011, 32, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Colasanti, A.; Owen, D.; Onega, M.; Kamalakaran, A.; Bennacef, I.; Matthews, P.M.; Rabiner, E.A.; Turkheimer, F.; Gunn, R.N. Quantification of the Specific Translocator Protein Signal of 18F-PBR111 in Healthy Humans: A Genetic Polymorphism Effect on In Vivo Binding. J. Nucl. Med. 2013, 54, 1915–1923. [Google Scholar] [CrossRef] [Green Version]
- Lavisse, S.; Guillermier, M.; Herard, A.-S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Rémy, P.; Dollé, F.; et al. Reactive Astrocytes Overexpress TSPO and Are Detected by TSPO Positron Emission Tomography Imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef]
- Janssen, B.; Vugts, D.J.; Windhorst, A.D.; Mach, R.H. PET Imaging of Microglial Activation—Beyond Targeting TSPO. Molecules 2018, 23, 607. [Google Scholar] [CrossRef] [Green Version]
- Horti, A.; Naik, R.; Foss, C.A.; Minn, I.; Misheneva, V.; Du, Y.; Wang, Y.; Mathews, W.B.; Wu, Y.; Hall, A.; et al. PET imaging of microglia by targeting macrophage colony-stimulating factor 1 receptor (CSF1R). Proc. Natl. Acad. Sci. USA 2019, 116, 1686–1691. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Nishimura, T.; Kondo, H.; Ikeda, K.; Hayashi, Y.; McGeer, P.L. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 1994, 639, 171–174. [Google Scholar] [CrossRef]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.; Tang, T.M.; Lue, L.-F. Studies on Colony Stimulating Factor Receptor-1 and Ligands Colony Stimulating Factor-1 and Interleukin-34 in Alzheimer’s Disease Brains and Human Microglia. Front. Aging Neurosci. 2017, 9, 244. [Google Scholar] [CrossRef]
- Dawson, V.L.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Gullipalli, D.; Song, W.-C. Modeling complement-driven diseases in transgenic mice: Values and limitations. Immunobiology 2016, 221, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef]
- Burns, T.C.; Li, M.D.; Mehta, S.; Awad, A.J.; Morgan, A.A. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: A systematic bioinformatics-based critique of preclinical models. Eur. J. Pharmacol. 2015, 759, 101–117. [Google Scholar] [CrossRef]
- Healy, L.; Perron, G.; Won, S.-Y.; Rao, V.T.S.; Guiot, M.-C.; Moore, C.; Bar-Or, A.; Antel, J.P. Differential transcriptional response profiles in human myeloid cell populations. Clin. Immunol. 2018, 189, 63–74. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.e8. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Jaenisch, R. Nuclear reprogramming and pluripotency. Nature 2006, 441, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Jesudoss, M.X.D.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Bahmad, H.F.; Hadadeh, O.; Chamaa, F.; Cheaito, K.; Darwish, B.; Makkawi, A.-K.; Abou-Kheir, W. Modeling Human Neurological and Neurodegenerative Diseases: From Induced Pluripotent Stem Cells to Neuronal Differentiation and its Applications in Neurotrauma. Front. Mol. Neurosci. 2017, 10, 1276. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals that Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Gomez-Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.; Nguyen, C.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.; Fimbres, C.; et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, A.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Hasselmann, J.; Coburn, M.A.; England, W.; Velez, D.X.F.; Shabestari, S.K.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, M.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Battin, C.; Hennig, A.; Mayrhofer, P.; Kunert, R.; Zlabinger, G.J.; Steinberger, P.; Paster, W. A human monocytic NF-κB fluorescent reporter cell line for detection of microbial contaminants in biological samples. PLoS ONE 2017, 12, e0178220. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Manivannan, J.; Teh, D.B.L.; Chua, S.M.; Gharibani, P.; All, A.H. A review of induced pluripotent stem cell, direct conversion by trans-differentiation, direct reprogramming and oligodendrocyte differentiation. Regen. Med. 2016, 11, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Llerena, C.V.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Möller, T.; Boddeke, E.; Prinz, M. Central nervous system myeloid cells as drug targets: Current status and translational challenges. Nat. Rev. Drug Discov. 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Solfrizzi, V.; Panza, F. Are NSAIDs Useful to Treat Alzheimer’s Disease or Mild Cognitive Impairment? Front. Aging Neurosci. 2010, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M.; et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12-week, double-blind, placebo-controlled study. Curr. Alzheimer Res. 2013, 10, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.L.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2017, 66, 1200–1212. [Google Scholar] [CrossRef]
- Josselyn, S.A. The past, present and future of light-gated ion channels and optogenetics. eLife 2018, 7. [Google Scholar] [CrossRef]
- Campbell, E.J.; Marchant, N.J. The use of chemogenetics in behavioural neuroscience: Receptor variants, targeting approaches and caveats. Br. J. Pharmacol. 2018, 175, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Hirbec, H.; Déglon, N.; Foo, L.C.; Goshen, I.; Grutzendler, J.; Hangen, E.; Kreisel, T.; Linck, N.; Muffat, J.; Regio, S.; et al. Emerging technologies to study glial cells. Glia 2020. [Google Scholar] [CrossRef] [PubMed]




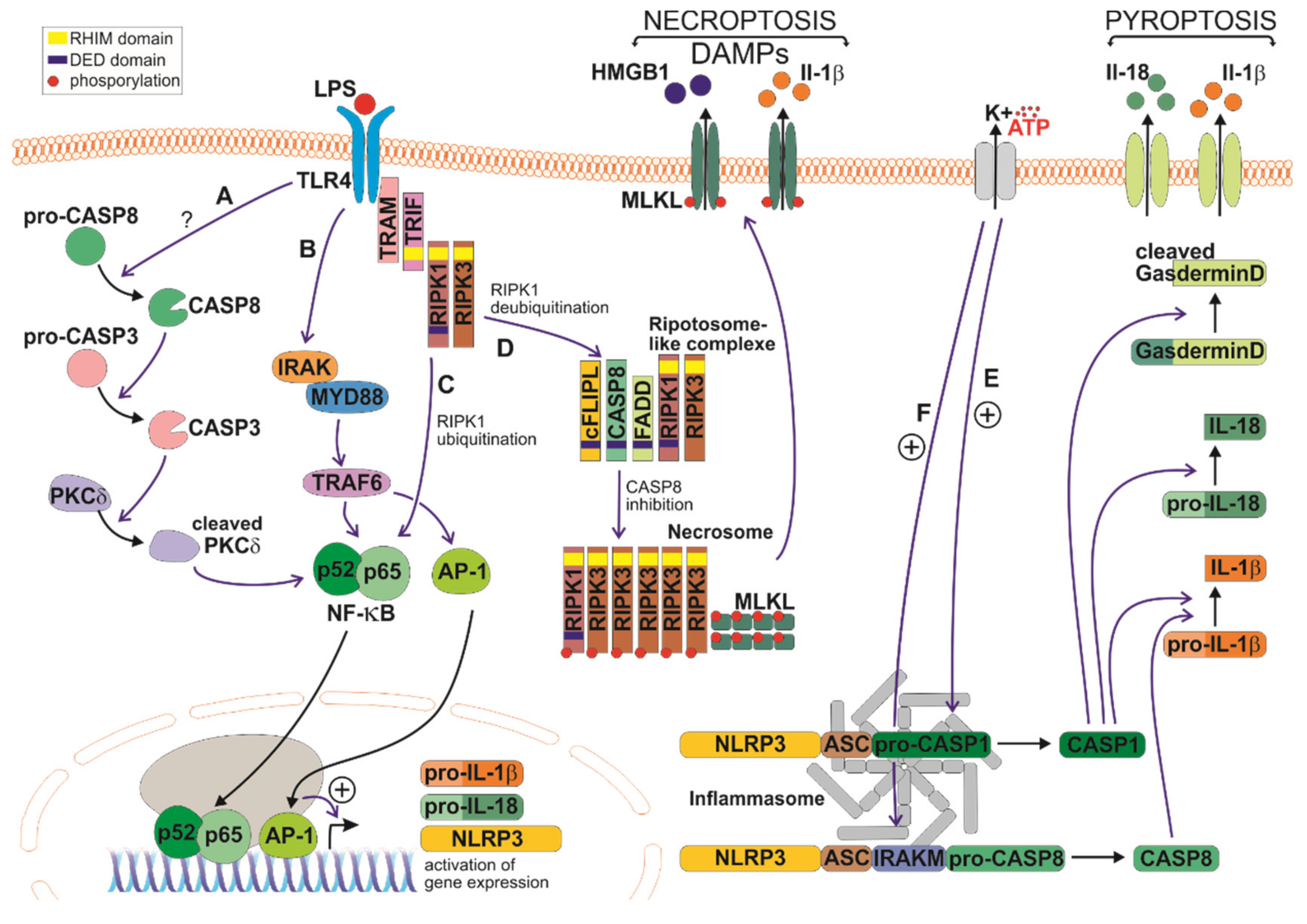{kind=link}
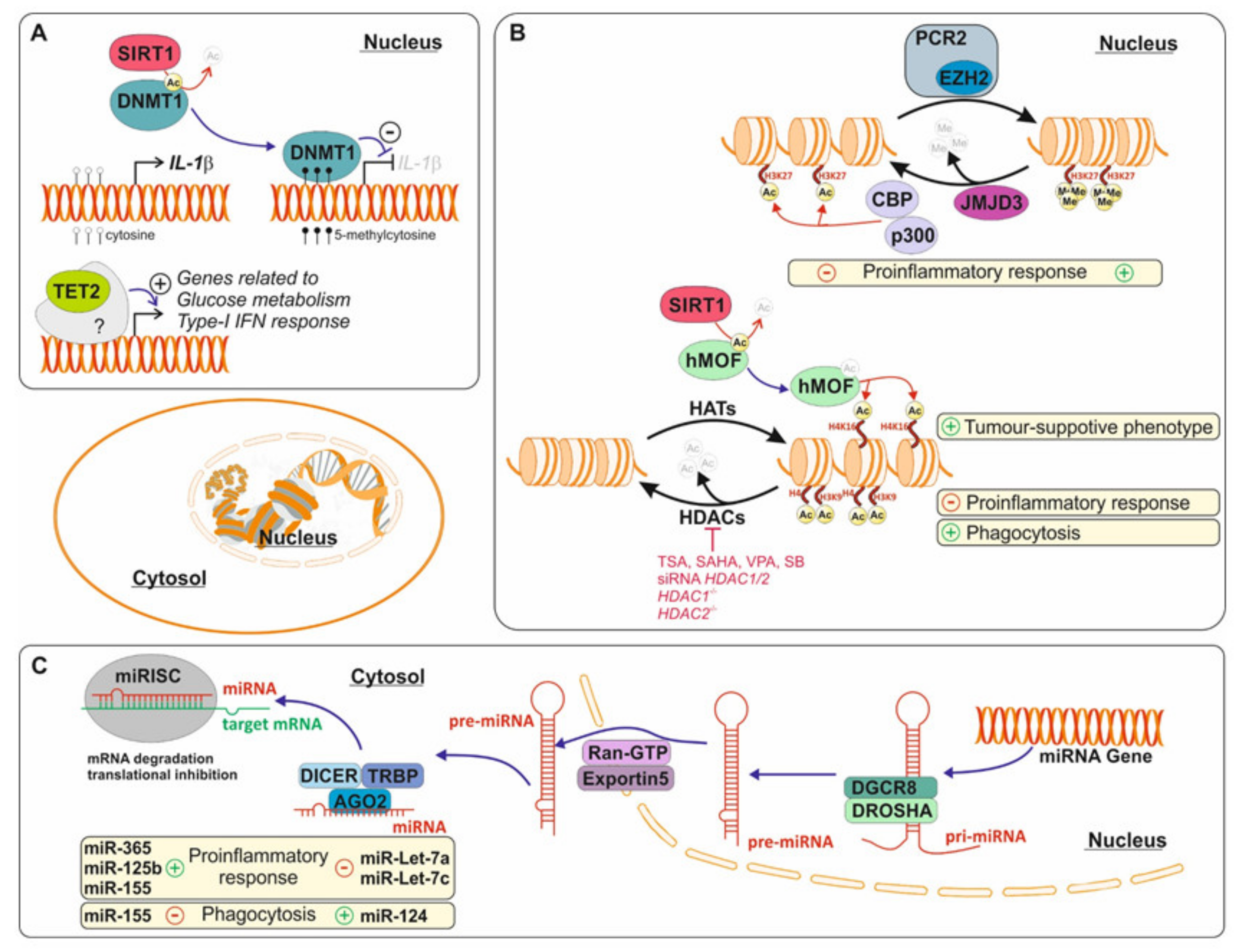{kind=link}
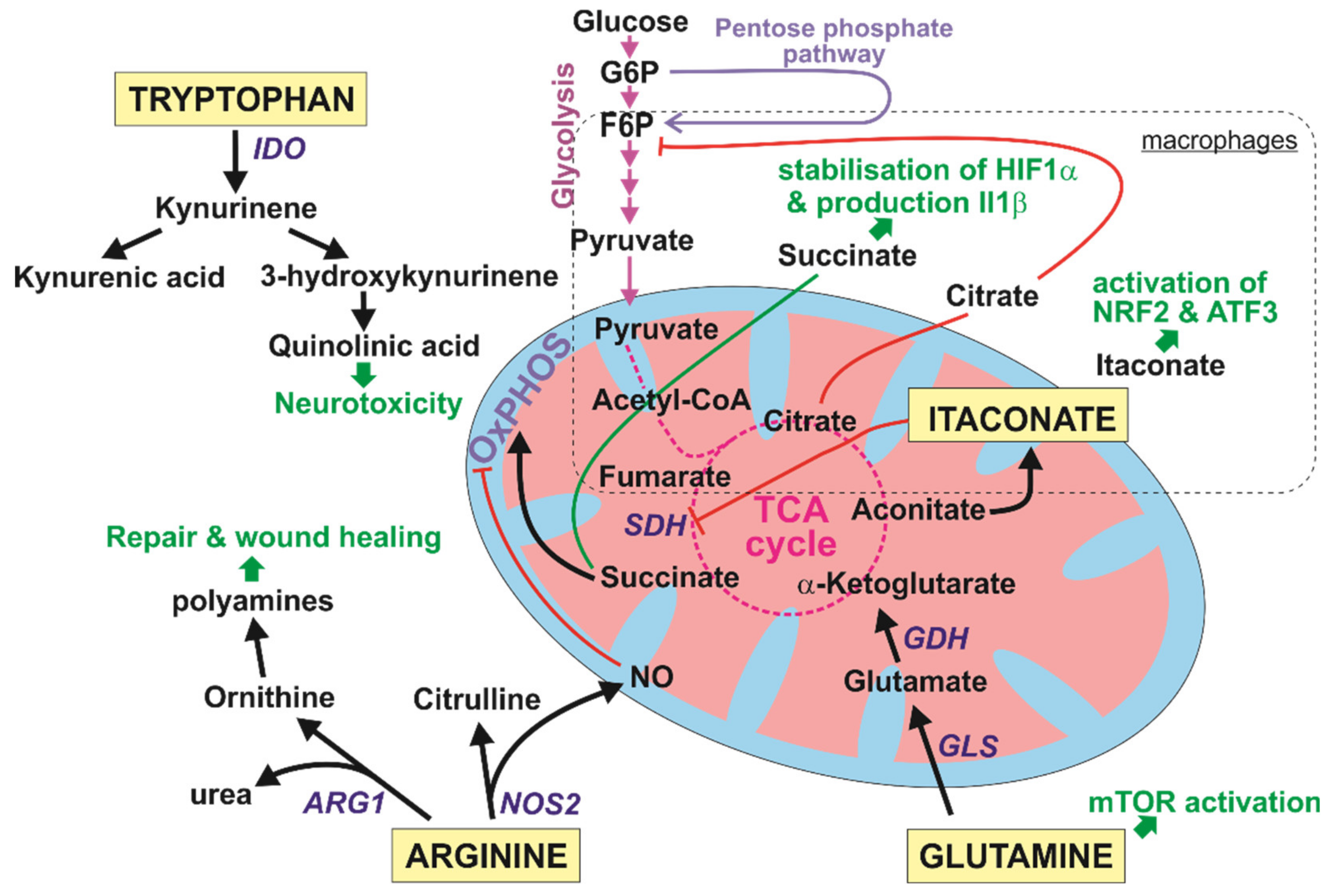{kind=link}
| TLR | Expression Level | PAMPs | DAMPs | |
|---|---|---|---|---|
| M | H | |||
| 1 | + | +++ | Lipoproteins [67] | α-syn [68] |
| 2 | +++++ | ++++ | PG [67,69], lipoproteins, LTA, zymosan [67], synthetic bacterial lipopeptide Pam3CysSK4 [70] | α-syn [68,71], Aβ [72], gangliosides, hyaluronic acid [73] |
| 3 | + | ++ | Poly(I:C), viral dsRNA [67,74] | mRNA from apoptotic cells [75], stathmin[76] |
| 4 | + | ++ | LPS [77], monophosphoryl lipid A [78] | Aβ [79], α-syn [80,81], MPP+ [82], HSP60, fibrinogen [73] |
| 5 | + | + | Bacterial flagellin [67] | ND |
| 6 | + | PG [69], lipoproteins, LTA [67] | HMGB1 [83] | |
| 7 | ++++ | ++ | Loxoribine [84], miR [85], ssRNA [67,86] | Self RNA, microRNA [83] |
| 8 | + | + | ssRNA [67] | Self RNA, microRNA [83] |
| 9 | ++++ | + | CpG-DNA [87,88], CpG-ODN [84,89], bacterial DNA [67] | DNA degenerating neurons [90], HMGB1 [91] |
| 10 | NF | + | ND | ND |
| 11 | + | NF | Profilin [92] | ND |
| 12 | + | NF | ND | ND |
| 13 | ++ | NF | Bacterial RNA [93] | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. https://doi.org/10.3390/cells9071717
Rodríguez-Gómez JA, Kavanagh E, Engskog-Vlachos P, Engskog MKR, Herrera AJ, Espinosa-Oliva AM, Joseph B, Hajji N, Venero JL, Burguillos MA. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells. 2020; 9(7):1717. https://doi.org/10.3390/cells9071717
Chicago/Turabian StyleRodríguez-Gómez, José A., Edel Kavanagh, Pinelopi Engskog-Vlachos, Mikael K.R. Engskog, Antonio J. Herrera, Ana M. Espinosa-Oliva, Bertrand Joseph, Nabil Hajji, José L. Venero, and Miguel A. Burguillos. 2020. "Microglia: Agents of the CNS Pro-Inflammatory Response" Cells 9, no. 7: 1717. https://doi.org/10.3390/cells9071717