
Evaluation of the Clinical Utility of Genomic Profiling to Inform Selection of Clinical Trial Therapy in Salivary Gland Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Consent and Clinical Data Collection
2.2. Next-Generation Sequencing
2.3. Systematic Evaluation of Genomic Biomarker and Drug Trial Matches
2.4. Classification of Level of Evidence
3. Results
3.1. Patient Characteristics
3.2. Next-Generation Sequencing with 24-Gene Targeted Panel
3.2.1. Systematic Evaluation of Matched Drug Therapies within Clinical Trials
3.2.2. TP53
3.2.3. PIK3CA/AKT/PTEN
3.2.4. Receptor Tyrosine Kinases—ERBB2 and EGFR
3.2.5. Others—BRAF, KRAS and CTNNB1
3.3. Biomarker-Matched Trial Availability
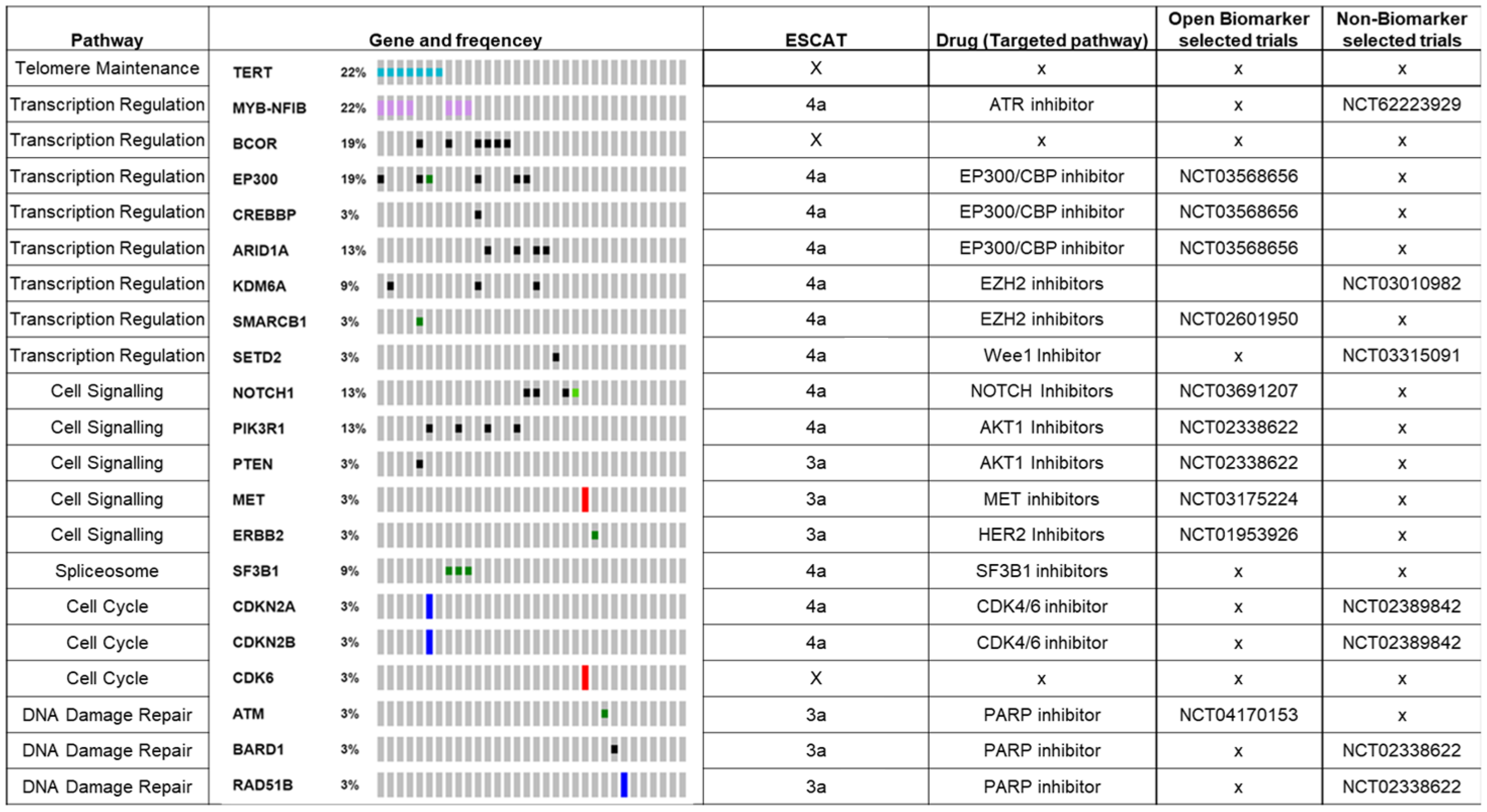
3.4. Next-Generation Sequencing with 350+ Gene Targeted Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, H.H.; Limesand, K.H.; Ann, D.K. Current State of Knowledge on Salivary Gland Cancers. Crit. Rev. Oncog. 2018, 23, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Di Villeneuve, L.; Souza, I.L.; Tolentino, F.D.S.; Ferrarotto, R.; Schvartsman, G. Salivary Gland Carcinoma: Novel Targets to Overcome Treatment Resistance in Advanced Disease. Front. Oncol. 2020, 10, 2097. [Google Scholar] [CrossRef] [PubMed]
- Chintakuntlawar, A.V.; Okuno, S.H.; Price, K.A. Systemic Therapy for Recurrent or Metastatic Salivary Gland Malignancies. Cancers Head Neck 2016, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; Bae, J.E.; Lorch, J.H.; Haddad, R.I.; Jo, V.Y.; Schoenfeld, J.D.; Margalit, D.N.; Tishler, R.B.; Goguen, L.A.; Annino, D.J., Jr.; et al. The Benefits of Adjuvant Trastuzumab for HER-2-Positive Salivary Gland Cancers. Oncology 2020, 25, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viscuse, P.V.; Price, K.A.; Garcia, J.J.; Schembri-Wismayer, D.J.; Chintakuntlawar, A.V. First Line Androgen Deprivation Therapy vs. Chemotherapy for Patients With Androgen Receptor Positive Recurrent or Metastatic Salivary Gland Carcinoma—A Retrospective Study. Front. Oncol. 2019, 9, 701. [Google Scholar] [CrossRef] [Green Version]
- Fushimi, C.; Tada, Y.; Takahashi, H.; Nagao, T.; Ojiri, H.; Masubuchi, T.; Matsuki, T.; Miura, K.; Kawakita, D.; Hirai, H.; et al. A Prospective Phase II Study of Combined Androgen Blockade in Patients with Androgen Receptor-Positive Metastatic or Locally Advanced Unresectable Salivary Gland Carcinoma. Ann. Oncol. 2018, 29, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Foster, N.R.; Zoroufy, A.J.; Worden, F.P.; Price, K.A.R.; Adkins, D.; Bowles, D.W.; Kang, H.; Burtness, B.; Sherman, E.J.; et al. Alliance A091404: A Phase II Study of Enzalutamide (NSC# 766085) for Patients with Androgen Receptor-Positive Salivary Cancers. J. Clin. Oncol. 2019, 37, 6020. [Google Scholar] [CrossRef]
- Li, B.T.; Shen, R.; Offin, M.; Buonocore, D.J.; Myers, M.L.; Venkatesh, A.; Razavi, P.; Ginsberg, M.S.; Ulaner, G.A.; Solit, D.B.; et al. Ado-Trastuzumab Emtansine in Patients with HER2 Amplified Salivary Gland Cancers (SGCs): Results from a Phase II Basket Trial. J. Clin. Oncol. 2019, 37, 6001. [Google Scholar] [CrossRef]
- Takahashi, H.; Tada, Y.; Saotome, T.; Akazawa, K.; Ojiri, H.; Fushimi, C.; Masubuchi, T.; Matsuki, T.; Tani, K.; Osamura, R.Y.; et al. Phase II Trial of Trastuzumab and Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2–Positive Salivary Duct Carcinoma. J. Clin. Oncol. 2018, 37, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Bowles, D.W.; Kang, H.; Meric-Bernstam, F.; Hainsworth, J.; Spigel, D.R.; Bose, R.; Burris, H.; Sweeney, C.J.; Beattie, M.S.; et al. Targeted Therapy for Advanced Salivary Gland Carcinoma Based on Molecular Profiling: Results from MyPathway, a Phase IIa Multiple Basket Study. Ann. Oncol. 2020, 31, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uijen, M.J.M.; Lassche, G.; van Engen-van Grunsven, A.C.H.; Tada, Y.; Verhaegh, G.W.; Schalken, J.A.; Driessen, C.M.L.; van Herpen, C.M.L. Systemic Therapy in the Management of Recurrent or Metastatic Salivary Duct Carcinoma: A Systematic Review. Cancer Treat. Rev. 2020, 89. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Haroon Al Rasheed, M.R.; Antonescu, C.R.; Alex, D.; Frosina, D.; Ghossein, R.; Jungbluth, A.A.; Katabi, N. Pan-Trk Immunohistochemistry Is a Sensitive and Specific Ancillary Tool for Diagnosing Secretory Carcinoma of the Salivary Gland and Detecting ETV6-NTRK3 Fusion. Histopathology 2020, 76, 375–382. [Google Scholar] [CrossRef]
- Kato, S.; Elkin, S.K.; Schwaederle, M.; Tomson, B.N.; Helsten, T.; Carter, J.L.; Kurzrock, R. Genomic Landscape of Salivary Gland Tumors. Oncotarget 2015, 6, 25631–25645. [Google Scholar] [CrossRef] [Green Version]
- Garrett, A.; Callaway, A.; Durkie, M.; Cubuk, C.; Alikian, M.; Burghel, G.J.; Robinson, R.; Izatt, L.; Talukdar, S.; Side, L.; et al. Cancer Variant Interpretation Group UK (CanVIG-UK): An Exemplar National Subspecialty Multidisciplinary Network. J. Med. Genet. 2020, 57, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. JMD 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and Validation of a Clinical Cancer Genomic Profiling Test Based on Massively Parallel DNA Sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.-Y.; et al. A Framework to Rank Genomic Alterations as Targets for Cancer Precision Medicine: The ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Marcinow, A.; Ozer, E.; Teknos, T.; Wei, L.; Hurtuk, A.; Old, M.; Agrawal, A.; Carrau, R.; Iwenofu, O.H. Clinicopathologic Predictors of Recurrence and Overall Survival in Adenoid Cystic Carcinoma of the Head and Neck: A Single Institutional Experience at a Tertiary Care Center. Head Neck 2014, 36, 1705–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinsky, K.; Hong, F.; McCourt, C.K.; Sachdev, J.C.; Mitchell, E.P.; Zwiebel, J.A.; Doyle, L.A.; McShane, L.M.; Li, S.; Gray, R.J.; et al. Effect of Capivasertib in Patients with an AKT1 E17K-Mutated Tumor: NCI-MATCH Subprotocol EAY131-Y Nonrandomized Trial. JAMA Oncol. 2021, 7, 271–278. [Google Scholar] [CrossRef]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical Relevance of Androgen Receptor Alterations in Prostate Cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.-C.; Soo, R.A.; Riely, G.J.; Ou, S.-H.I.; Clancy, J.S.; Li, S.; et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef]
- Dankner, M.; Lajoie, M.; Moldoveanu, D.; Nguyen, T.-T.; Savage, P.; Rajkumar, S.; Huang, X.; Lvova, M.; Protopopov, A.; Vuzman, D.; et al. Dual MAPK Inhibition Is an Effective Therapeutic Strategy for a Subset of Class II BRAF Mutant Melanomas. Clin. Cancer Res. 2018, 24, 6483. [Google Scholar] [CrossRef] [Green Version]
- Zaman, G.J.R.; de Roos, J.A.D.M.; Libouban, M.A.A.; Prinsen, M.B.W.; de Man, J.; Buijsman, R.C.; Uitdehaag, J.C.M. TTK Inhibitors as a Targeted Therapy for CTNNB1 Mutant Cancers. Mol. Cancer Ther. 2017, 16, 2609. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Tan, L.; Beauchamp, E.M.; Hatcher, J.M.; Liu, Q.; Meyerson, M.; Gray, N.S.; Hammerman, P.S. Characterization of DDR2 Inhibitors for the Treatment of DDR2 Mutated Nonsmall Cell Lung Cancer. ACS Chem. Biol. 2015, 10, 2687–2696. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2019, 382, 41–50. [Google Scholar] [CrossRef]
- Weaver, A.; Bossaer, J.B. Fibroblast Growth Factor Receptor (FGFR) Inhibitors: A Review of a Novel Therapeutic Class. J. Oncol. Pharm. Pract. 2020, 27, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma With IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Dogra, R.; Bhatia, R.; Shankar, R.; Bansal, P.; Rawal, R.K. Enasidenib: First Mutant IDH2 Inhibitor for the Treatment of Refractory and Relapsed Acute Myeloid Leukemia. Anti-Cancer Agents Med. Chem. 2018, 18, 1936–1951. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Rankin, C.; Blanke, C.D.; Demetri, G.D.; Borden, E.C.; Ryan, C.W.; von Mehren, M.; Blackstein, M.E.; Priebat, D.A.; Tap, W.D.; et al. Correlation of Long-Term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017, 3, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET -Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Ho, A.S.; Ochoa, A.; Jayakumaran, G.; Zehir, A.; Valero Mayor, C.; Tepe, J.; Makarov, V.; Dalin, M.G.; He, J.; Bailey, M.; et al. Genetic Hallmarks of Recurrent/Metastatic Adenoid Cystic Carcinoma. J. Clin. Investig. 2019, 129, 4276–4289. [Google Scholar] [CrossRef]
- De Bono, J.; de Giorgi, U.; Massard, C.; Bracarda, S.; Nava Rodrigues, D.; Kocak, I.; Font, A.; Arranz Arija, J.; Shih, K.; Radavoi, G.; et al. PTEN Loss as a Predictive Biomarker for the Akt Inhibitor Ipatasertib Combined with Abiraterone Acetate in Patients with Metastatic Castration-Resistant Prostate Cancer (MCRPC). Ann. Oncol. 2016, 27, vi243. [Google Scholar] [CrossRef]
- Klubo-Gwiezdzinska, J. Targeting RET-Mutated Thyroid and Lung Cancer in the Personalised Medicine Era. Lancet Diabetes Endocrinol. 2021, 9, 473–474. [Google Scholar] [CrossRef]
- Zhao, R.-X.; Xu, Z.-X. Targeting the LKB1 Tumor Suppressor. Curr. Drug Targets 2014, 15, 32–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective Killing of ATM- or P53-Deficient Cancer Cells through Inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Sheth, H.; Kumar, P.; Shreenivas, A.; Sambath, J.; Pragya, R.; Madre, C.; Athikari, N.; Khandare, H.; Peshattiwar, V.; Datar, R.; et al. Excellent Response With Alpelisib and Bicalutamide for Advanced Salivary Duct Carcinoma With PIK3CA Mutation and High Androgen Receptor Expression—A Case Report. JCO Precis. Oncol. 2021, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kim, C.; Kim, H.; Lee, C.; Lee, H.; Bae, W.; Jeung, H.; Zang, D.; Park, S.; Hwang, I.; et al. SO-10 An Open-Label, Multi-Centre, Phase Ib/II Study of PI3Kβ Selective Inhibitor GSK2636771 Administered in Combination with Paclitaxel in Patients with Advanced Gastric Cancer Having Alterations in PI3K/Akt Pathway. Ann. Oncol. 2021, 32, s206. [Google Scholar] [CrossRef]
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J. Clin. Oncol. 2018, 36, 2532–2537. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants Are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; et al. ATR Is a MYB Regulated Gene and Potential Therapeutic Target in Adenoid Cystic Carcinoma. Oncogenesis 2020, 9, 5. [Google Scholar] [CrossRef]
- Crabb, S.; Plummer, R.; Greystoke, A.; Carter, L.; Pacey, S.; Walter, H.; Coyle, V.M.; Knurowski, T.; Clegg, K.; Ashby, F.; et al. 560TiP A Phase I/IIa Study to Evaluate the Safety and Efficacy of CCS1477, a First in Clinic Inhibitor of P300/CBP, as Monotherapy in Patients with Selected Molecular Alterations. Ann. Oncol. 2021, 32, S617. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Wirth, L.J.; Muzaffar, J.; Rodriguez, C.P.; Xia, B.; Perez, C.A.; Bowles, D.W.; Winquist, E.; Hotte, S.J.; Metcalf, R.; et al. 919MO ACCURACY a Phase II Trial of AL101, a Selective Gamma Secretase Inhibitor, in Subjects with Recurrent/Metastatic (R/M) Adenoid Cystic Carcinoma (ACC) Harboring Notch Activating Mutations (Notchmut). Ann. Oncol. 2020, 31, S663. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.-C.; Zalmas, L.-P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by DNTP Starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Hall, T.; Eathiraj, S.; Wick, M.J.; Schwartz, B.; Abbadessa, G. In-Vitro and in-Vivo Combined Effect of ARQ 092, an AKT Inhibitor, with ARQ 087, a FGFR Inhibitor. Anti-Cancer Drugs 2017, 28, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Stahel, R.; Bubendorf, L.; Bonomi, P.; Villegas, A.; Kowalski, D.M.; Baik, C.S.; Isla, D.; Carpeno, J.D.C.; Garrido, P.; et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin. Cancer Res. 2019, 25, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The Biological Function and Clinical Significance of SF3B1 Mutations in Cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef]
- Min, A.; Kim, Y.J.; Lee, M.; Lee, K.-H.; Im, S.-A. Abstract PS5-40: CDKN2A Loss Can Be a Predictive Marker of Palbociclib in Breast and Gastric Cancer. Cancer Res. 2021, 81, PS5-40. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in Patients with Cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Bowles, D.W.; Even, C.; Hao, D.; Kang, H.; Metcalf, R.; Muzaffar, J.; Oliva, M.; Perez, C.A.; Popovtzer, A.; et al. 904P ACCURACY: A Phase II Trial of AL101, a Selective Gamma Secretase Inhibitor, in Subjects with Recurrent/Metastatic (R/M) Adenoid Cystic Carcinoma (ACC) Harboring Notch Activating Mutations (Notchmut): Results of 6-Mg Cohort. Ann. Oncol. 2021, 32, S803–S804. [Google Scholar] [CrossRef]
- Adderley, H.; Rack, S.; Hapuarachi, B.; Feeney, L.; Morgan, D.; Hussell, T.; Wallace, A.J.; Betts, G.; Hodgson, C.; Harrington, K.; et al. The Utility of TP53 and PIK3CA Mutations as Prognostic Biomarkers in Salivary Adenoid Cystic Carcinoma. Oral Oncol. 2021, 113, 105095. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Li, Q.; Xia, Y.; Chan, C.-C.; Yuan, X.; Wang, Y.; Chen, S.; Qian, W. Preclinical Evaluation of SC0191, a Small Molecule Inhibitor of Wee1 Kinase. J. Clin. Oncol. 2020, 38, e15637. [Google Scholar] [CrossRef]
- Chen, A.P.; Kummar, S.; Moore, N.; Rubinstein, L.V.; Zhao, Y.; Williams, P.M.; Palmisano, A.; Sims, D.; O’Sullivan Coyne, G.; Rosenberger, C.L.; et al. Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT): A Randomized Multicenter Phase II Trial. JCO Precis. Oncol. 2021, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. JOURNAL OF CLINICAL ONCOLOGY Phosphatidylinositol 3-Kinase a-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Mitani, Y.; Diao, L.; Guijarro, I.; Wang, J.; Zweidler-McKay, P.; Bell, D.; William, W.N., Jr.; Glisson, B.S.; Wick, M.J.; et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J. Clin. Oncol. 2017, 35, 352–360. [Google Scholar] [CrossRef]
- Hapuarachi, B.S.; Feeney, L.; Rack, S.; Adderley, H.; Morgan, D.; Betts, G.; Walker, R.; Rauch, R.; Herz, E.; Harrington, K.J.; et al. Clinical Disease Course and Survival Outcomes Following Disease Recurrence in Adenoid Cystic Carcinoma (ACC) with NOTCH Signaling Pathway Activation. J. Clin. Oncol. 2021, 39, 6072. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.H.; Lee, J.; Choi, M.; Cho, J.; Kim, S.J.; Kim, W.S.; Ko, Y.H.; Yoo, H.Y. The Mutation of BCOR Is Highly Recurrent and Oncogenic in Mature T-Cell Lymphoma. BMC Cancer 2021, 21, 82. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of CtDNA to Support Patient Selection for Early Phase Clinical Trials: The TARGET Study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Liu, S.; Fu, K.; Gao, N.; Li, R.; He, W.; Gao, Z. Clinicopathological Characteristics and Outcomes of 23 Patients with Secretory Carcinoma of Major Salivary Glands. Sci. Rep. 2021, 11, 22639. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristic | Subgroup | Total | NGS Successful | NGS Failed | |||
|---|---|---|---|---|---|---|---|
| Number | Percentage | Number | Percentage | Number | Percentage | ||
| All Patients | 209 | 100 | 175 | 84 | 34 | 16 | |
| Sex | Female | 113 | 54 | 93 | 53 | 20 | 59 |
| Male | 96 | 46 | 82 | 47 | 14 | 41 | |
| Age | Median | 51 | 51 | 51 | |||
| Range | 13–78 | 13–77 | 23–78 | ||||
| Site | Minor | 93 | 44 | 75 | 43 | 18 | 53 |
| Major | 116 | 56 | 100 | 57 | 16 | 47 | |
| R/M disease | Yes | 190 | 91 | 159 | 91 | 31 | 91 |
| No | 19 | 9 | 16 | 9 | 3 | 9 | |
| Metastatic sites | Local | 78 | 37 | 56 | 32 | 22 | 65 |
| Lung | 121 | 58 | 99 | 57 | 22 | 65 | |
| Bone | 37 | 18 | 32 | 18 | 5 | 15 | |
| Liver | 22 | 11 | 22 | 13 | 0 | 0 | |
| Other | 24 | 11 | 21 | 12 | 3 | 9 | |
| Subtypes | ACC | 142 | 68 | 115 | 66 | 27 | 79 |
| Adeno | 18 | 9 | 15 | 9 | 3 | 9 | |
| SDC | 17 | 8 | 16 | 9 | 1 | 3 | |
| Acinic | 9 | 4 | 8 | 5 | 1 | 3 | |
| ExPleo | 8 | 4 | 8 | 5 | 0 | 0 | |
| MEC | 7 | 3 | 6 | 3 | 1 | 3 | |
| MyoE | 5 | 2 | 4 | 2 | 1 | 3 | |
| NEC | 1 | 1 | 1 | 1 | 0 | 0 | |
| Secretory | 1 | 1 | 1 | 1 | 0 | 0 | |
| NUT | 1 | 1 | 1 | 1 | 0 | 0 | |
| GENE | ESCAT | Open Biomarker Selected Trials | Drug (Targeted Pathway) |
|---|---|---|---|
| AKT1 | 3a [24] | NCT03673787 | Ipatasertib (AKT1 inhibitor) |
| NCT01226316, NCT02338622 | AZD5363 (AKT1 inhibitor) | ||
| AR | 4a [25] | No trial | - |
| ALK | 3a [26] | No trial | - |
| BRAF | 3a [27] | NCT02407509 | RO5126766 (Raf/MEK inhibitor) |
| CTNNB1 | 4a [28] | No trial | - |
| DDR2 | 4a [29] | No trial | - |
| EGFR | 3a [30] | NCT04259450 | AFM24 (anti-EGFR antibody) |
| ERBB2 | 3a/3b [6,10] | NCT01953926 | Neratinib (Pan-Her inhibitor) |
| NCT03410927 | TAS0728 (Her-2 inhibitor) | ||
| FGFR2 | 4a [31] | NCT02052778, NCT04189445 | Futibatinib (FGFR inhibitor) |
| NCT04083976 | Erdafitinib (FGFR inhibitor) | ||
| NCT03822117 | Pemigatinib (FGFR inhibitor) | ||
| GNA11 | 4a [32] | No trial | - |
| GNAQ | 4a [32] | No trial | - |
| IDH1 | 3a [33] | NCT03684811 | Olutasidenib (IDH1 inhibitor) |
| IDH2 | 3a [34] | No trial | - |
| KIT | 3a [35] | NCT02571036 | Ripretinib (KIT/PGDRa inhibitor) |
| KRAS | 4a [27,36] | NCT02407509 | RO5126766 (Raf/MEK inhibitor) |
| MAP2K1 | 3a [36] | NCT02407509 | RO5126766 (Raf/MEK inhibitor) |
| MET | 3a [37] | NCT02925104 | Capmatinib (MET inhibitor) |
| NRAS | 3a [36] | NCT02407509 | RO5126766 (Raf/MEK inhibitor) |
| PDGFRa | 3a [35] | NCT02508532 | Avapritinib (PDGFRa inhibitor) |
| PIK3CA | 2b [38] | NCT01226316, NCT02338622 | AZD5363 (AKT1 inhibitor) |
| NCT03006172 | GDC-0077 (Pi3K inhibitor) | ||
| PTEN | 3a [39] | NCT03673787 | Ipatasertib (AKT1 inhibitor) |
| NCT01226316, NCT02338622 | AZD5363 (AKT1 inhibitor) | ||
| RET | 3a [40] | NCT03037385 | Pralsetinib (RET inhibitor) |
| NCT03157128 | Selpercatinib (RET inhibitor) | ||
| STK11 | 4a [41] | No trial | - |
| TP53 | 4a [42] | No trial | - |
| Patient | Subtype | Variant | Patient | Subtype | Variant |
|---|---|---|---|---|---|
| ACC1 | ACC | TP53 c.832C > T p.(Pro278Ser) 17% reads | ACC16 | ACC | PIK3CA c.1633G > A p.(Glu545Lys) 11% reads |
| ACC3 | ACC | TP53 c.814G > T p.(Val272Leu) 4% reads | ACC19 | ACC | PIK3CA c.1258T > C p.(Cys420Arg) 18% reads; |
| ACC10 | ACC | TP53 c.328delC p.(Arg110ValfsTer13) 6% reads | ACC19 | ACC | PIK3CA c.1633G > A p.(Glu545Lys) 8% reads |
| ACC13 | ACC | TP53 c.1146delA p.(Lys382AsnfsTer40) 29% reads | ACINIC25 | ACINIC | PIK3CA c.3140A > G p.(His1047Arg) 23% reads |
| ACC14 | ACC | TP53 c.375G > A p.(Thr125Thr) 28% reads | ADENO32 | ADENO | PIK3CA c.1636C > A p.(Gln546Lys) 36.41% reads |
| ACC15 | ACC | TP53 c.584T > C p.(Ile195Thr) 7% reads | ExPleo35 | ExPleo | PIK3CA c.1633G > A p.(Glu545Lys) 39% reads |
| ACC18 | ACC | TP53 c.373A > G p.(Thr125Ala) 14% reads | SDC44 | SDC | PIK3CA c.1624G > A p.(Glu542Lys) 13% reads |
| ACC19 | ACC | PIK3CA c.1633G > A p.(Glu545Lys) 8% reads | SDC45 | SDC | PIK3CA c.1093G > A p.(Glu365Lys) 20% reads |
| ACC21 | ACC | TP53 c.329G > C p.(Arg110Pro) 50% reads | SDC46 | SDC | PIK3CA c.3140A > G p.(His1047Arg) 13% reads |
| ACC22 | ACC | TP53 c.824G > A p.(Cys275Tyr) 4% reads | SDC47 | SDC | PIK3CA c.1624G > A p.(Glu542Lys) 13% reads |
| ACC22 | ACC | TP53 c.467G > C p.(Arg156Pro) 8% read | SDC49 | SDC | PIK3CA c.3140A > G p.(His1047Arg) 10% reads |
| ACC24 | ACC | TP53 c.1010G > A p.(Arg337His) 13% reads | SDC52 | SDC | PIK3CA c.3140A > G p.(His1047Arg) 51% reads |
| ACINIC25 | ACINIC | TP53 c.742C > T p.(Arg248Trp) 4% reads | ACC1 | ACC | AKT1 c.49G > A p.(Glu17Lys) 4% reads |
| ACINIC26 | ACINIC | TP53 c.818G > T p.(Arg273Leu) 35% reads | SDC54 | SDC | AKT1 c.49G > A p.(Glu17Lys) 22% reads |
| ADENO27 | ADENO | TP53 c.794T > G p.(Leu265Arg) 37% reads | SDC56 | SDC | AKT1 c.49G > A p.(Glu17Lys) 19% reads |
| ADENO28 | ADENO | TP53 c.637C > T p.(Arg213Ter) 26% reads | ACC5 | ACC | PTEN c.829dup p.(Thr277AsnfsTer21) 9% reads |
| ADENO29 | ADENO | TP53 c.626_627delGA p.(Arg209LysfsTer6) 30% reads | ACC7 | ACC | PTEN c.750_765dup p.(Glu256TrpfsTer2) 52% reads |
| ADENO30 | ADENO | TP53 c.481G > A p.(Ala161Thr) 22% reads; | ACC12 | ACC | PTEN c.16A > T p.(Lys6Ter) 62% reads |
| ExPleo34 | ExPleo | TP53 c.686_687dupGT p.(Thr230ValfsTer18) 38% reads | ACC17 | ACC | PTEN c.686C > G p.(Ser229Ter) 12% reads |
| ExPleo35 | ExPleo | TP53 c.991C > T p.(Gln331Ter) 44% reads; | ADENO31 | ADENO | PTEN c.382A > C p.(Lys128Gln) 7% reads |
| ExPleo36 | ExPleo | TP53 c.750_754dupCATCC p.(Leu252ArgfsTer95) 20% reads | MEC40 | MEC | PTEN c.634 + 5G > T 42% reads |
| ExPleo37 | ExPleo | TP53 c.989T > G p.(Leu330Arg) 26% reads | ACC4 | ACC | EGFR c.2369C > T p.(Thr790Met) 31% reads |
| MEC38 | MEC | TP53 c.828delC p.(Cys277ValfsTer68) 7% reads | ExPleo36 | ExPleo | ERBB2 c.2305G > C p.(Asp769His) 81% reads |
| MEC39 | MEC | TP53 c.733G > A p.(Gly245Ser) 15% reads | SDC43 | SDC | ERBB2 c.929C > T p.(Ser310Phe) 34% reads |
| MEC41 | MEC | TP53 c.460_462delGGC p.(Gly154del) 60% reads | SDC50 | SDC | ERBB2 c.2305G > C p.(Asp769His) 26% reads |
| SDC42 | SDC | TP53 c.839G > C p.(Arg280Thr) 14% reads | ACC11 | ACC | BRAF c.1397G > A p.(Gly466Glu) 27% reads |
| SDC43 | SDC | TP53 c.993 + 1G > T 41% reads | SDC45 | SDC | BRAF c.1780G > A p.(Asp594Asn) 20% reads; |
| SDC45 | SDC | TP53 c.374C > G p.(Thr125Arg) 22% reads | ADENO27 | ADENO | KRAS c.436G > A p.(Ala146Thr) 8% reads |
| SDC48 | SDC | TP53 c.841G > C p.(Asp281His) 39% reads. | ACC6 | ACC | CTNNB1 c.134C > T p.(Ser45Phe) 23% reads |
| SDC50 | SDC | TP53 c.377_380delACTC p.(Tyr126SerfsTer43) 18% reads | |||
| SDC52 | SDC | TP53 c.626_627delGA p.(Arg209LysfsTer6) 43% reads | |||
| SDC53 | SDC | TP53 c.919 + 1G > C 25% reads | |||
| SDC54 | SDC | TP53 c.650T > G p.(Val217Gly) 15% reads | |||
| SDC55 | SDC | TP53 c.452C > G p.(Pro151Arg) 21% reads | |||
| SDC56 | SDC | TP53 c.912delT p.(Lys305SerfsTer40) 17% reads |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rack, S.; Feeney, L.; Hapuarachi, B.; Adderley, H.; Woodhouse, L.; Betts, G.; Burghel, G.J.; Harrington, K.J.; Metcalf, R. Evaluation of the Clinical Utility of Genomic Profiling to Inform Selection of Clinical Trial Therapy in Salivary Gland Cancer. Cancers 2022, 14, 1133. https://doi.org/10.3390/cancers14051133
Rack S, Feeney L, Hapuarachi B, Adderley H, Woodhouse L, Betts G, Burghel GJ, Harrington KJ, Metcalf R. Evaluation of the Clinical Utility of Genomic Profiling to Inform Selection of Clinical Trial Therapy in Salivary Gland Cancer. Cancers. 2022; 14(5):1133. https://doi.org/10.3390/cancers14051133
Chicago/Turabian StyleRack, Samuel, Laura Feeney, Brindley Hapuarachi, Helen Adderley, Laura Woodhouse, Guy Betts, George J. Burghel, Kevin J. Harrington, and Robert Metcalf. 2022. "Evaluation of the Clinical Utility of Genomic Profiling to Inform Selection of Clinical Trial Therapy in Salivary Gland Cancer" Cancers 14, no. 5: 1133. https://doi.org/10.3390/cancers14051133