
Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate

1
Institute for Organic Chemistry (IOC), RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
2
Department of Chemistry, University of Jyvaskyla, P.O. Box 35, 40014 Jyväskylä, Finland
*
Author to whom correspondence should be addressed.
Molbank 2023, 2023(3), M1710; https://doi.org/10.3390/M1710
Submission received: 27 July 2023
/
Revised: 2 August 2023
/
Accepted: 5 August 2023
/
Published: 9 August 2023
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Site-selective functionalization of pyridines is a crucial tool for the synthesis of diverse pharmaceuticals and materials. We introduced diiminium pyridine adducts as highly convenient and potent Lewis acids. We report that tributylphosphine selectively adds to the 4-position of pyridine in tetramethyldiiminium pyridine ditrifluoromethanesulfonate, resulting in the formation of the title compound. This finding represents an advancement towards the utilization of diiminium units as organic reagents or catalysts for pyridine functionalization. We also employ computational models to determine fluoride and hydride ion affinities, Fukui function f+(r), molecular electrostatic potential, and pKa values, providing valuable insights for future investigations in this area.
1. Introduction
Pyridines play a crucial role in pharmaceutical and chemical applications [1]. However, achieving site-selective functionalization of pyridines remains a challenging task [2,3,4]. N-functionalization of pyridines can induce C2- and C4-selectivity, with bulkier N-substituents favoring the latter [5,6,7,8,9,10,11]. Addition to the C4-position leads to the formation of 1,4-dihydropyridine (DHP) structures, which can further undergo C3-functionalization [12,13,14,15]. The synthesis of substituted 1,4-DHPs was historically challenging [16,17,18,19], leading to recent advancements towards addressing this issue effectively. Pyridines substituted at the C2-position with bulky groups, for example, were found to form “frustrated” Lewis pairs (FLPs) with boranes, enabling complete reduction with molecular hydrogen [20,21,22,23,24,25]. Partial reduction can also be achieved through hydroboration and hydrosilyzation strategies [26,27,28,29].
In our recent work, we investigated three diiminium nucleophile adducts, including the pyridine adduct (DIPy) 1, as carbon-based Lewis acids (Figure 1) [30]. These adducts offer a cost-efficient and convenient synthesis method at large scales and proved to be potent coupling reagents for amide and imide formations. The adducts typically engage in a “double-action” mechanism involving the addition of a Lewis base and subsequent elimination of the pyridines. This behavior resembles that of Lewis pairs and complicates direct comparison of their acidity with simple single-action Lewis acids. However, we determined that they exhibit characteristics consistent with hard and soft Lewis super-acids [31]. We successfully abstracted fluoride ions from SbF6−, albeit under harsh conditions, following the typical double-action mechanism. Oxygen nucleophiles displayed similar reactivity. Interestingly, when employing the soft hydride donor Et3SiH, we observed behavior more in line with the established chemistry of N-substituted pyridines, with the C4-position of the diiminium adduct acting as a better soft Lewis acid than the diiminium carbon. Our regiospecific synthesis of the 1,4-DHP 3 sparked our interest in employing diiminium nucleophile adducts for pyridine substitution. Consequently, we initiated investigations with heavier and more complex soft bases, and herein describe the addition of tributylphosphine to diiminium pyridine adduct 1 and examine the acidity properties of the resulting product. While they are important intermediates in the functionalization of pyridines, only six phosphonium adducts of 1,4-DHP have been isolated [32,33].
2. Results and Discussion
2.1. Synthesis
Preliminary experiments were conducted on a 250 μmol scale using dichloromethane (DCM) and diethyl ether (Et2O) in an argon atmosphere in screw-capped 10 mL vials. In 1.5 mL of the respective solvent, 1.00 eq. of 1 and 1.05 eq. of PBu3 were mixed and stirred for 2 h at room temperature (r.t.) under argon. The solvent was evaporated, and the reaction progress was evaluated using 1H nuclear magnetic resonance (NMR) in CD3CN. The remaining pyridine integrals of 1 compared to the vinylic integrals of the product tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium ditrifluoromethanesulfonate 4 suggested 88% completion in DCM and 81% completion in Et2O (assuming a specific reaction). Based on these results, we chose to run the synthesis overnight (18 h) to ensure complete conversion.
2.50 mmol of DIPy 1 were reacted with 2.65 mmol (1.05 eq.) of PBu3 in 10 mL anhydrous Et2O under an argon atmosphere (Figure 2). The dispersion slowly turned pale-yellow. The solid crude product was collected after 18 h in air by filtration on a paper filter and washed with Et2O. It was then dissolved in DCM and filtrated. Removal of the solvent yielded the title compound 4 at satisfactory purity in an excellent yield (97%). Traces of 1 and [Bu3POPBu3]OTf2 were be detected in 1H and 31P NMR spectra, respectively. The latter was identified by analogy of the 31P NMR shift (87 ppm) to the known shift of [Et3POPEt3]OTf2. (87 ppm) [30]. A high purity sample was obtained by recrystallisation in an argon atmosphere rather than filtration in air. To achieve this, DCM was added to the solution of another reaction after 18 h until the product dissolved. The title compound crystallized at −18 °C and was collected in the form of crystalline needles. Combined with further crystallization from the mother liquor, pure 4 was isolated in 92% yield. Storage of 4 in a dry atmosphere is advised due to mild hygroscopy.
Analogous reactions with the poorer nucleophiles triphenylphosphine and triphenylphosphite did not lead to isolable adducts. No reaction was observed with triphenylphosphite. Triphenylphosphine led to the formation of sets of broad 1H NMR signals indicative of the desired 1,4-DHP but we were able to reisolate both starting materials through washing with Et2O, which dissolved the phosphine. This suggests that triphenylphosphine reversible adds to DIPy 1, which may be useful for promoting other functionalization reactions.
2.2. Structure in the Solid State
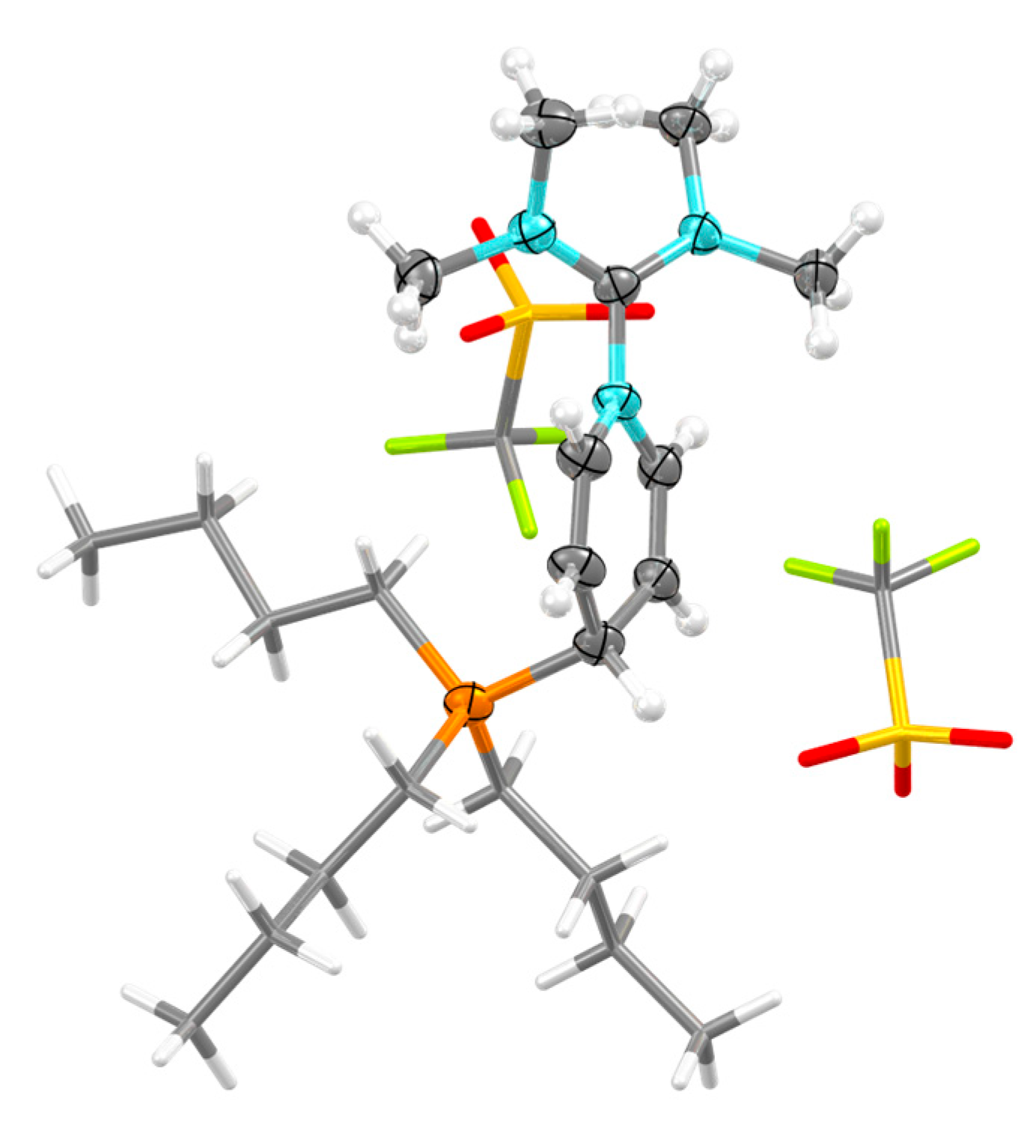
Crystals suitable for structure determination by single crystal X-ray diffraction (SCXRD) of compound 4 were generated by recrystallisation from Et2O/DCM at −18 °C (see synthesis section for details). The colorless plates were mounted on MiTeGen 100 µm micro-mounts using a non-polar cryo-oil for subsequent determination by single crystal X-ray crystallography (Figure 3, not simplified in Figure S13). The asymmetric cell contained one molecule of 4. One triflate anion was found to be disordered over two positions in a 60%:40% ratio and was modelled accordingly.
The bond length of 137.2(3) pm indicates a strong bond in the diiminium-C–dihydropyridyl-N bond. In comparison, the diiminium-C–pyridine-N bond in 1 is 144.8(2) pm long [30]. The P–C–C bond angles at the dihydropyridyl-C4 position are 109.5(2) and 111.2(2)°, consistent with an sp3-carbon center. The respective P–C bond length measures 183.2(3) pm.
The diiminium carbon exhibits a distance of 318.2(3) pm to the closest triflate oxygen. On the other side of the diiminium carbon, there is a disordered triflate anion from a neighboring unit cell. The distances to the two closest disordered oxygens of this triflate measure 312.6(8) pm and 323.6(6) pm, with an average of 317 pm (with the individual distances weighted by the 60%:40% ratio of the triflates). These distances are smaller than the sum of the van der Waals radii (322 pm) and suggest a weak Lewis adduct formation at the diiminium carbon.
2.3. Nuclear Magnetic Resonance
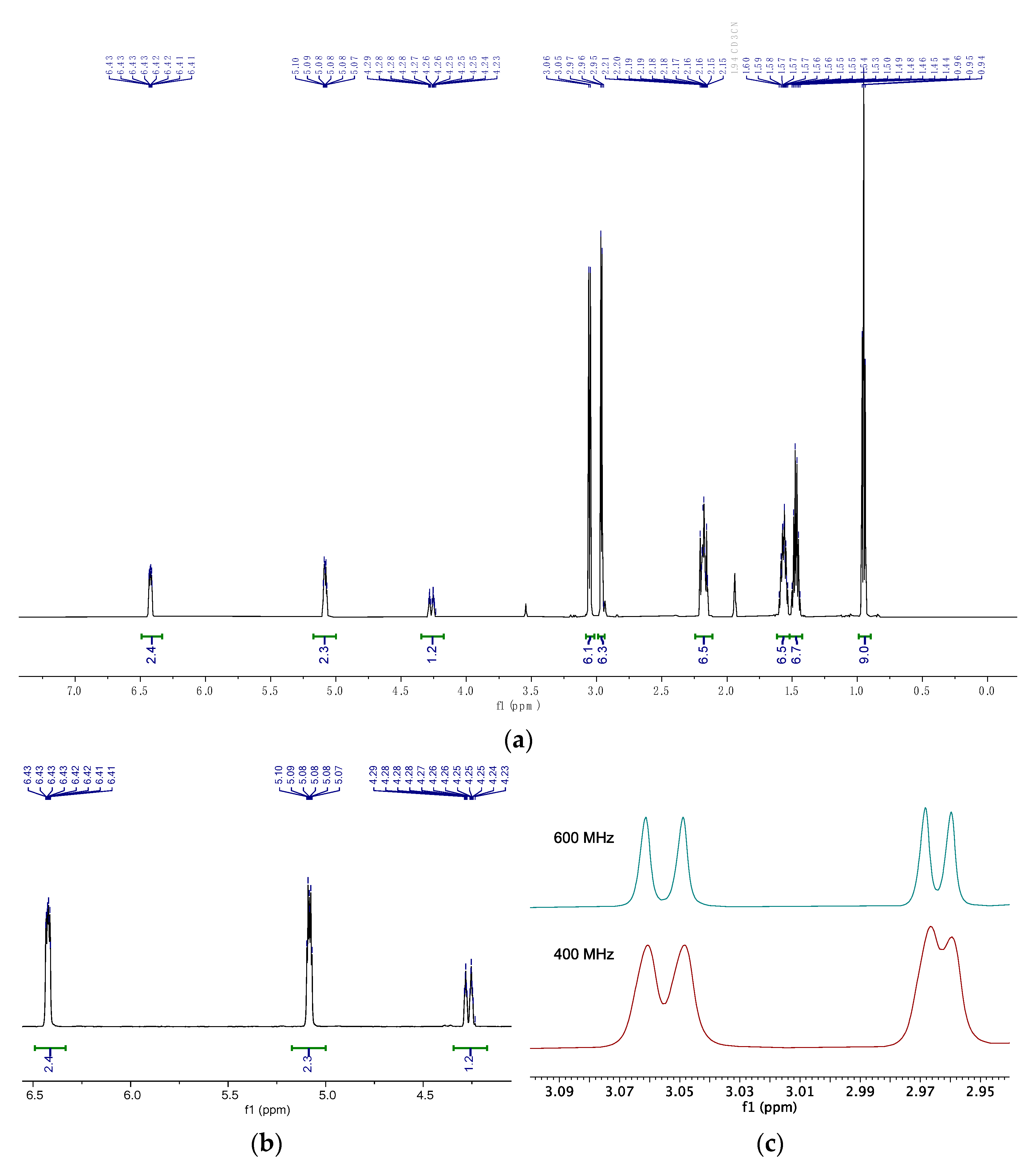
Analysis by nuclear magnetic resonance (NMR) spectroscopy revealed intriguing details (Figure 4a, also see Figures S1–S12). The proton in DHP-position 4 displayed a relatively high coupling constant to P (2J(HP) = 18.5 Hz), while positions 3 and 2 exhibited coupling constants of 3J(HP) = 3.0 and 4J(HP) = 3.8 Hz, respectively (Figure 4b). These findings were consistent with the scarce previous reports on similar phosphonium-substituted DHPs [32]. Interestingly, we detected a total of four iminium-methyl signals both in 1H and 13C NMR spectra, with no evidence of HH couplings by 1H/1H COSY NMR. The major splitting can be attributed to frozen rotations of the iminium bonds (which is also known for compound 1) [30]. To explore the origin of this observation, we compared 1H NMR spectra at different field strengths (600 and 400 MHz), and the constant shifts (in parts per million, ppm) eliminated coupling as an explanation for the minor line-splitting (Figure 4c). This suggests that the diiminium unit remains immobilized relative to the DHP plane at r.t. on the 1H and 13C NMR timescale. The discovery that small methyl substituents are sufficient to freeze this degree of freedom makes the development of enantioselective follow-up chemistry with unsymmetric derivatives of 4 promising.
2.4. Computational Investigation
To facilitate further rational investigations, we modelled various properties of 4. Calculations were performed at the DSD-BLYP-D3BJ/def2-QZVPP/CPCM(MeCN)//PBEh-3c/def2-mSVP/CPCM(MeCN) level of theory [34,35,36,37,38,39,40,41,42,43]. The structure of 4 was obtained and pre-optimized using a pipeline described in detail in the computational methods section. We modelled the fluoride and hydride ion affinities (FIA/HIA) both in the gas phase and in MeCN solution (FIAMeCN/HIAMeCN) to evaluate the hard and soft acidity of 4 (Figure 5). To avoid direct calculations of hydride and fluoride ions, we employed Krossing’s anchoring scheme with Greb’s Me3Si+ anchor [44,45,46,47,48]. This scheme employs the FIA and HIA of the Me3Si+ at the level used herein: FIA = 963 kJ mol−1; HIA = 962 kJ mol−1. These were anchored to a high level coupled cluster method as described previously [30]. The solvation energy of fluoride and hydride ions was used to determine ion affinities in solution: ΔHsolv(F−) = −370 kJ mol−1; ΔHsolv(H−) = −368 kJ mol−1. The FIA of the diiminium carbon in 4 was determined to be 779 kJ mol−1, while the HIA was found to be 956 kJ mol−1. These values indicate significantly lower acidities compared to 1 (FIA: 983 kJ mol−1, HIA: 1153 kJ mol−1) [30]. The phosphonium unit, acting as an electrophilic phosphonium cation [49], displayed an FIA of 773 kJ mol−1 and an HIA of 801 kJ mol−1, indicating weaker acidity than the carbon but with more balanced acidities towards soft and hard bases. Dicationic Lewis acids experience strong stabilization by solvation. Modelling the polar solvent MeCN resulted in substantial acidity dampening, with all values dropping below 25 kJ mol−1, except for the HIA of the diiminium unit, which remained significant at 181 kJ mol−1.
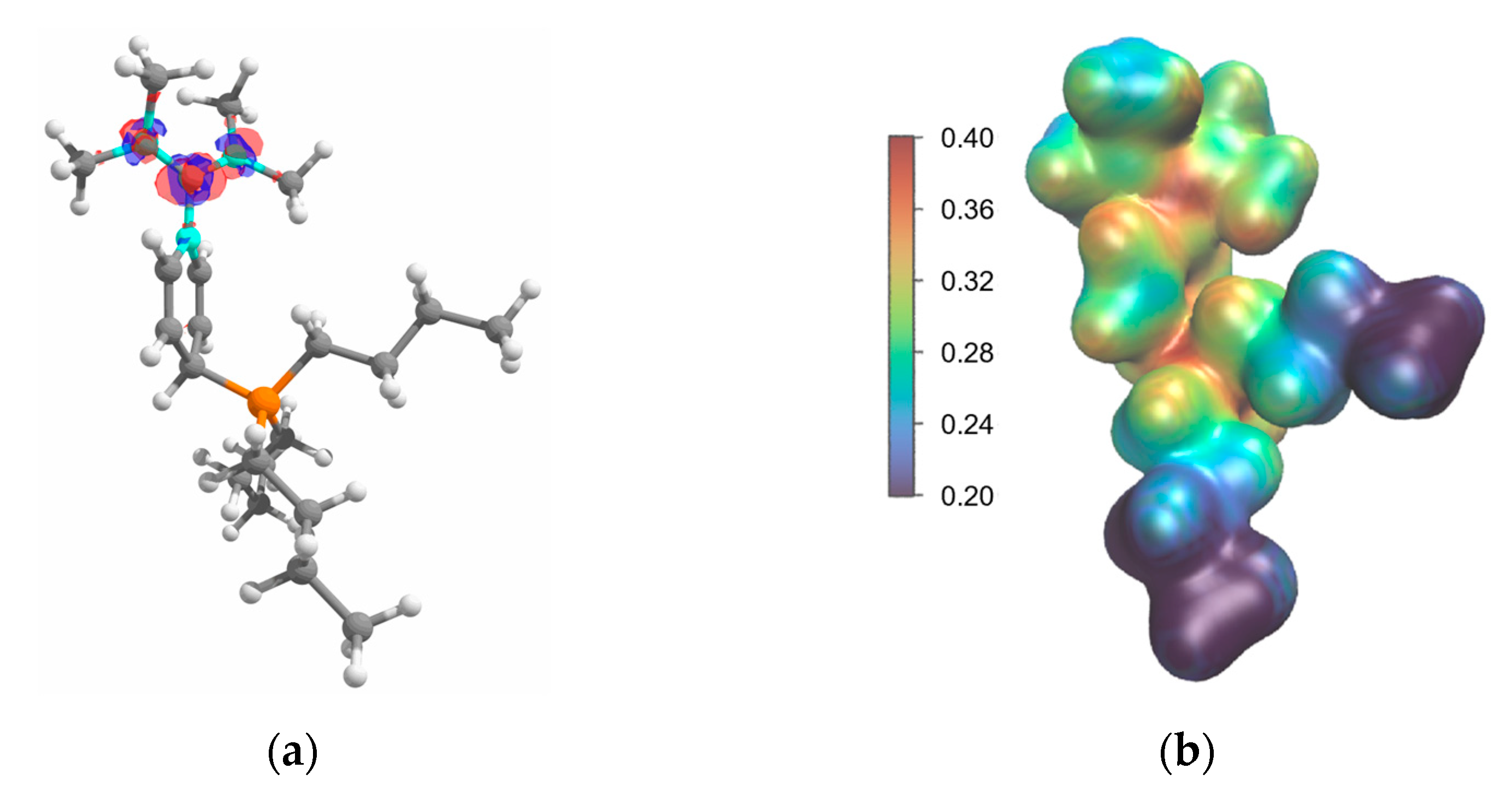
As previously observed, molecular electrostatic potentials (MEPs) provide valuable predictions of nucleophilic attacks at 1 and its derivatives under thermodynamic reaction control, while Fukui functions align with outcomes under kinetic control [30]. No clear distinction between the diiminium carbon and the phosphonium phosphorus centers was observed in the MEP. Both positions exhibit similarly strong positive electrostatic potentials which aligns well with their similar FIA values (Figure 6a). The Fukui function f+(r) clearly indicates that the most electrophilic position is the diiminium carbon atom (Figure 6b). Minor coefficients are visible at the iminium nitrogen. DIPy 1 showed the largest coefficients in the pyridine ring, which is in line with the observation of the phosphine addition to the pyridine C4-position.
pKa values were obtained by modeling deprotonations with the reference base 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) using Equation (1):
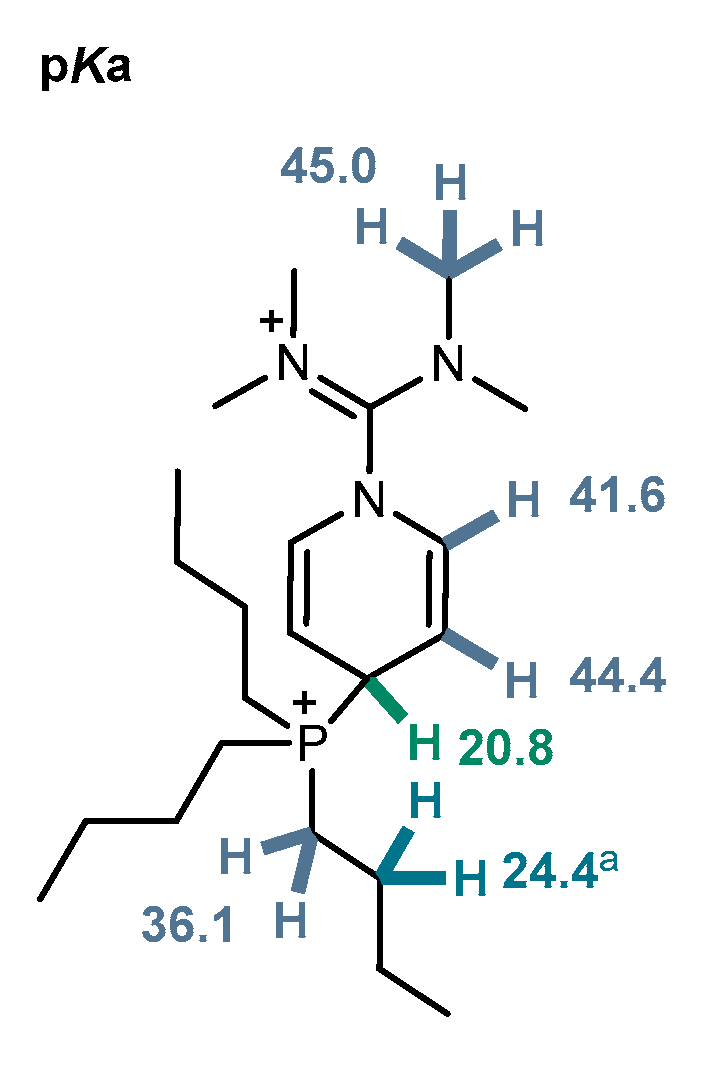
with and (Figure 7) [50,51]. The iminium-methyl groups and the vinylic protons of the DHP unit were predicted to have pKa values of 42–45. Deprotonation of the methylene groups neighboring phosphorus to form an ylide was only slightly more favorable, with a pKa of 36. While we were not able to compute a pure deprotonation at the butyls, the Hoffmann-type elimination of butene under β-CH2-deprotonation was modeled to proceed with a pKa of 24. The most acidic position predicted was the C4-position of the DHP, with a pKa of 21. This can be attributed to the formation of a conjugated system extending from the resulting ylide through the heterocycle to the iminium units. Subsequent elimination of the phosphine to yield a carbene at its former position is as unlikely (218 kJ mol−1 uphill) as spontaneous elimination of phosphonium HPBu3+ without external base (229 kJ mol−1 uphill).
3. Materials and Methods
3.1. Experimental Methods
All synthetic procedures were carried out in an argon-filled glovebox (O2 and H2O < 3 ppm) or in Schlenk-type glassware in a dry argon atmosphere unless stated otherwise. Glassware used in these procedures was pre-heated in an oven at 120 °C for at least 2 h and flame-dried with a heat gun and flushed with dry argon thrice.
All chemicals were obtained from commercial suppliers and used without further purification unless stated otherwise. Dichloromethane (DCM) and diethyl-ether (Et2O) were dried with an MBRAUN solvent purification system (SPS-5) and stored over 4 Å molecular sieves. Deuterated solvents for anhydrous measurements were introduced to a glovebox as obtained from Eurisotop and stored over 4 Å molecular sieves. Cannula filtrations were performed with polytetrafluoroethylene (PTFE) tubing capped with MN615 filter paper from MACHEREY-NAGEL (retention capacity 4–12 μm) attached with PTFE tape.
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance Neo 600 (Bruker, Billerica, USA) spectrometer at 26 °C and were analysed with the software Mnova 14.2.3 [52]. Chemical shifts δ were reported in ppm. The residual solvent signals were used for referencing 1H NMR spectra (1.94 ppm for CD3CN) [53,54]. This shift was used for absolute referencing of all other spectra with the unified chemical shifts scale as recommended by the International Union of Pure and Applied Chemistry (IUPAC) in its implementation in Mnova 14.2.3 (13C: Me4Si j = 1%, X = 25.145020; 19F: CCl3F X = 94.094011; 31P: H3PO4 external, X = 40.480742) [52,55]. Multiplicities of the signals were given as singlet (s), doublet (d), triplet (t), quartet (q), quintet (qui), septet (sept), multiplet (m), or broad signal (bs). Elemental analyses (CHN) were carried out in air with an Elementar VarioEL (Elementar, Langenselbold, Germany) or a Bruker maXis II (Bruker, Billerica, United States). High-resolution mass spectrometry (HRMS, further abbreviated as MS) was performed using a Thermo Scientific LTQ Orbitrap XL (Thermo Fisher Scientific, Waltham, USA). Attenuated total reflection infrared spectra (ATR-IR) were measured on a PerkinElmer Spectrum 100 FT-IR (PerkinElmer, Waltham, United States) using the universal ATR (UATR) accessory and the signals were reported in cm−1. Single-crystal X-ray data was measured using a Rigaku XtaLAB Synergy-R (Rigaku, Tokyo, Japan) diffractometer with a HyPix-Arc 100 detector using mirror-monochromated Cu-Kα (λ = 1.54184 Å) radiation. The data collection and reduction were performed using the program CrysAlisPro 1.171.42.89a [56]. The structures were solved with intrinsic phasing (SHELXT) [57] and refined by full-matrix least squares on F2 using Olex2 1.2 [58], which utilizes the SHELXL module [59]. Anisotropic displacement parameters were assigned to non-H atoms. All hydrogen atoms were refined using riding models with Ueq(H) of 1.5Ueq(C) for methyl groups and Ueq(H) of 1.2Ueq(C) for all other C-H groups.
3.2. Computational Methods
Calculations were performed in autodE 1.3.0 [60]. Low energy conformers were generated from simplified molecular-input line-entry system (SMILES) tags with the ETKDGv3 algorithm implemented in RDKit v. 2022.03.4 [61]. Conformer optimizations were performed using xTB 6.4.0 [62] at the GFN2-xTB level [63]. Solution phase calculations used the generalized Born solvation model with a hydrophobic solvent accessible surface area (GBSA), with parameters appropriate for MeCN [64,65]. Feasibly low energy conformers as selected with autodE default settings were then optimized with ORCA 5.0.4 [66,67,68] with the triple-corrected composite method PBEh-3c/def2-mSVP [39,40,41,42] utilising the RIJCOSX [43] approximation for Coulomb and Hartree Fock exchange with the def2/J auxiliary basis [36]. autodE’s template-based algorithm including conformer screening was used for the localization of most transition states. Relaxed potential energy scans as implemented in ORCA were used for localizing rotational transition states, which were then optimized and further treated within autodE. Hessians were then computed in ORCA at the same level of theory for confirming the nature of the optimized structures as local minimum or maximum, respectively. autodE was used to compute the zero-point energy and free energy corrections based on the generated Hessian. The frequencies were scaled by 0.95 as advised for this functional and basis set combination [39]. Low frequency vibration modes were treated with the quasi-rigid-rotor-harmonic-oscillator approximation (qRRHO, with ω0 = 100 cm−1) [69]. Potential imaginary frequencies smaller than 40 cm−1 were not considered as transition states and were treated as real frequencies within thermochemistry calculations (multiplication by −i). Electronic energies were obtained from single point calculations at the DSD-BLYP [34] level with D3BJ [40,41] dispersion correction with the basis set def2-QZVPP [35] and the auxiliary basis sets def2/J [36] and def2-QZVPP/C [37]. Optimization, Hessian, and single point calculations in solution utilized the conductor-like polarizable continuum model (C-PCM) with parameters appropriate for MeCN, unless stated otherwise [38]. Energies were calculated at a 1 M concentration and a temperature of 25 °C, unless stated otherwise.
Compound energies are listed as Eopt, Gcont, Hcont, and Esp. Eopt is the energy of the final structure at our level of theory used for geometry optimization. Esp is the energy of the compound at our single point calculation level. Hcont is the enthalpy contribution, i.e., zero-point energy (H = Esp + Hcont). Gcont is the total free energy contribution, i.e., the sum of the zero-point energy and entropy contributions (G = Esp + Gcont).
The molecular electrostatic potential was computed with 80 grid intervals and projected onto the electron density surface at ρ = 0.01, utilizing ORCA 5.0.4 and a Python 3 script (used in Python 3.9.16) available at https://gist.github.com/mretegan/5501553 (revision of 25 Oct. 2022, accessed on 28. Apr. 2023). Fukui functions f+(r) were computed from single point calculations of the dications and the respective mono-cationic radicals and displayed at ρ = 0.01.
To assist with the editing process and improve the clarity and readability of the manuscript, an AI language model, ChatGPT (based on GPT-3.5 architecture) was employed. It provided suggestions and recommendations for improving the writing style and structure of the manuscript.
3.3. Synthesis of 4
2.50 mmol (1.19 g, 1.00 eq.) of DIPy 1 (synthesized according to [30]) were dissolved in 10 mL anhydrous Et2O in a 100 mL Schlenk tube equipped with a magnetic stir bar. Then, 2.63 mmol (669 µL, 1.05 eq.) of PBu3 were added dropwise under stirring. The reaction mixture was allowed to stir at r.t. for 18 h, resulting in the formation of a pale-yellow dispersion. Purification was performed following either of the following methods:
Purification procedure A: The reaction mixture was filtered swiftly in air with a paper filter. The collected solid was washed thrice using 10 mL of Et2O each time. Subsequently, the solid was dissolved in 10 mL of DCM and filtered through the paper filter. The solvent was evaporated from the filtrate in vacuum to yield 2.43 mmol (1.65 g, 97%) of compound 4.
Purification procedure B: To achieve a higher purity at a slightly reduced yield, crystallization in an argon atmosphere in the reaction Schlenk flask was performed. We added DCM (ca. 20 mL) via cannula to the reaction dispersion of compound 4 until it dissolved completely. The flask was tightly sealed and placed in a freezer at −18 °C. After 12 h, crystals were observed to form and were subsequently collected using a filter cannula. The mother liquor was collected in a second Schlenk flask and subjected to the same crystallization method again (5 mL Et2O, ca. 18 mL DCM) to yield further crystals. Both collected crystal batches were thoroughly washed with Et2O via filter cannula. A total of 2.31 mmol (1.57 g, 92%) of 4 were isolated as a pale-blue wool-like material consisting of crystalline needles.
1H NMR (600 MHz, CD3CN) δ 6.42 (dt, J = 8.3, 4.3, 1.5 Hz, 2H, DHP-2 CH), 5.08 (dt, J = 8.3, 3.8, 3.0 Hz, 2H, DHP-3 CH), 4.34–4.17 (dtd, J = 18.5, 3.8, 1.5 Hz, 1H, DHP-4 CH), 3.06 (s, 3H, Me-CH3), 3.05 (s, 3H, Me-CH3), 2.97 (s, 3H, Me-CH3), 2.96 (s, 3H, Me-CH3), 2.24–2.11 (m, 6H, Bu-CH2), 1.62–1.52 (m, 6H, Bu-CH2), 1.47 (tq, J = 7.3, 7.2 Hz, 6H, Bu-CH2), 0.95 (t, J = 7.3 Hz, 9H, Bu-CH3). 13C NMR (151 MHz, CD3CN) δ 157.5 (s, 1C, diiminium-C), 129.3 (d, J = 8.0 Hz, 2C, DHP-2 CH), 120.3 (q, J = 320.9 Hz, 2C, Tf-CF3), 98.6 (d, J = 6.3 Hz, 2C, DHP-3 CH), 40.1 (s, 1C NCH3), 40.0 (s, 1C NCH3), 39.5 (s, 1C NCH3), 39.4 (s, 1C, NCH3), 29.1 (d, J = 48.5 Hz, 1C, DHP-4 CH), 22.8 (d, J = 15.1 Hz, 3C, Bu-CH2), 22.2 (d, J = 4.9 Hz, 3C, Bu-CH2), 15.4 (d, J = 44.5 Hz, 3C, Bu-CH2), 11.7 (s, 3C, Bu-CH3). 31P NMR (243 MHz, CD3CN) δ 31.6 (1P). 19F NMR (565 MHz, CD3CN) δ −80.1 (6F, CF3). ATR-IR (neat) 3093, 2962, 2876, 2302, 2193, 2066, 1985, 1692, 1621, 1538, 1463, 1415, 1320, 1253, 1146, 1098, 1027, 985, 921, 894, 834, 912, 791, 760, 727. MS (ESI+) m/z 530.27776 (530.27876 calcd. for C23H44F3N3O3PS+ [4 − TfO−]+); 380.31830 (380.31891 calcd. for C22H43N3P+ [4 − 2 TfO− − H+]+); 280.21833 (280.21886 calcd. for C17H31NP+ [4 − 2 TfO− − Bu+ − CH4]+); 249.05113 (249.05152 calcd. for C6H12F3N2O3S+ [(Me2N)2COTf]+); 190.66270 (190.66309 calcd. for C22H44N3P2+ [4 − 2 TfO−]2+). EA (average of four determinations) C, 41.94; H, 6.36; N, 6.17 (C, 42.41; H, 6.53; N, 6.18 calcd. for C24H44F6N3O6PS2). Crystal data: CCDC-2283768, [C22H44N3P][CF3O3S]2, M = 679.71, colorless plate, 0.02 × 0.05 × 0.11 mm, triclinic, space group P-1 (No. 2), a = 10.2279(4) Å, b = 10.9774(6) Å, c = 15.1854(8) Å, α = 79.443(4)°, β = 84.043(4)°, γ = 81.475(4)°, V = 1652.42(14) Å3, Z = 2, Dcalc = 1.366 gcm3, F(000) = 716, µ = 2.58 mm1, T = 120.0(1) K, θmax = 74.5°, 6710 total reflections, 4802 with Io > 2σ(Io), Rint = 0.074, 6710 data, 459 parameters, 114 restraints, GooF = 1.05, 0.49 < d∆ρ < 0.52 eÅ3, R[F2 > 2σ(F2)] = 0.055, wR(F2) = 0.156.3.
4. Conclusions
Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium ditrifluoromethanesulfonate 4 was successfully synthesized through the addition of tributylphosphine to the diiminium pyridine adduct 1. Compound 4 contains both a strong carbon-based and a strong phosphorus-based Lewis acid, exhibiting comparable FIA and HIA values. The high computed pKa value at the C4 position of the dihydropyridine suggests its suitability for deprotonation or oxidative aromatization.
Supplementary Materials
The following supporting information can be downloaded online. This appendix contains the NMR spectra of compound 4 (Figures S1–S12) after purification with procedure B, unless stated otherwise. Figure S13. Structure of 4 determined by SCXRD.
Author Contributions
Conceptualization, F.F.M.; methodology, F.F.M., Y.G., J.S.W. and K.R.; validation, F.F.M., Y.G., J.S.W. and K.R.; formal analysis, F.F.M., Y.G., J.S.W. and K.R.; investigation, F.F.M., Y.G., J.S.W. and K.R.; resources, F.F.M. and K.R.; data curation, F.F.M., Y.G., J.S.W. and K.R.; writing—original draft preparation, F.F.M.; writing—review and editing, F.F.M., Y.G., J.S.W. and K.R.; visualization, F.F.M. and J.S.W.; supervision, F.F.M. and K.R.; project administration, F.F.M. and K.R.; funding acquisition, F.F.M. and K.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fonds der Chemischen Industrie, grant number Li 210/01 (F.F.M.) and by the Alexander von Humboldt Foundation through a Feodor Lynen Return Fellowship (F.F.M.) and through a Humboldt Research Award (K.R.). Computing resources were granted by the RWTH Aachen University under project rwth0928.
Data Availability Statement
Deposition number CCDC2283768 contains the supplementary crystallographic data for compound 4. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service. DOI 10.19061/iochem-bd-6-266 contains the supplementary computational data for compound 4 and the required derivatives. These data are provided free of charge in ioChem-BD hosted by the Barcelona Supercomputing Center (BSC).
Acknowledgments
Generous support was received through materials and infrastructure by Carsten Bolm, his group, and the Institute for Organic Chemistry at the RWTH Aachen University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Stephens, D.E.; Larionov, O.V. Recent Advances in the C-H-Functionalization of the Distal Positions in Pyridines and Quinolines. Tetrahedron 2015, 71, 8683–8716. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhou, F.-Y. Recent Developments in Transition-Metal-Free Functionalization and Derivatization Reactions of Pyridines. Synlett 2020, 32, 159–178. [Google Scholar] [CrossRef]
- Maity, S.; Bera, A.; Bhattacharjya, A.; Maity, P. C-H functionalization of pyridines. Org. Biomol. Chem. 2023, 21, 5671–5690. [Google Scholar] [CrossRef]
- Kim, M.; Koo, Y.; Hong, S. N-Functionalized Pyridinium Salts: A New Chapter for Site-Selective Pyridine C-H Functionalization via Radical-Based Processes under Visible Light Irradiation. Acc. Chem. Res. 2022, 55, 3043–3056. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Mathi, G.R.; Hong, S.; Hong, S. Enantioselective functionalization at the C4 position of pyridinium salts through NHC catalysis. Nat. Commun. 2022, 13, 1776. [Google Scholar] [CrossRef]
- Ma, X.; Herzon, S.B. Intermolecular Hydropyridylation of Unactivated Alkenes. J. Am. Chem. Soc. 2016, 138, 8718–8721. [Google Scholar] [CrossRef]
- Fier, P.S. A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J. Am. Chem. Soc. 2017, 139, 9499–9502. [Google Scholar] [CrossRef]
- Mathi, G.R.; Kweon, B.; Moon, Y.; Jeong, Y.; Hong, S. Regioselective C−H Functionalization of Heteroarene N-Oxides Enabled by a Traceless Nucleophile. Angew. Chem. Int. Ed. 2020, 59, 22675–22683. [Google Scholar] [CrossRef]
- Choi, J.; Laudadio, G.; Godineau, E.; Baran, P.S. Practical and Regioselective Synthesis of C-4-Alkylated Pyridines. J. Am. Chem. Soc. 2021, 143, 11927–11933. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Liu, Z.; He, J.-H.; Zhang, M.; Shi, Z.-J.; Tang, H.; Zhou, X.-Y.; Tian, J.-J.; Wang, X.-C. Borane-Catalyzed C3-Alkylation of Pyridines with Imines, Aldehydes, or Ketones as Electrophiles. J. Am. Chem. Soc. 2022, 144, 4810–4818. [Google Scholar] [CrossRef]
- Muta, R.; Torigoe, T.; Kuninobu, Y. 3-Position-Selective C–H Trifluoromethylation of Pyridine Rings Based on Nucleophilic Activation. Org. Lett. 2022, 24, 8218–8222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-Y.; Zhang, M.; Liu, Z.; He, J.-H.; Wang, X.-C. C3-Selective Trifluoromethylthiolation and Difluoromethylthiolation of Pyridines and Pyridine Drugs via Dihydropyridine Intermediates. J. Am. Chem. Soc. 2022, 144, 14463–14470. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, Z.-J.; Liu, L.; Zhang, M.; Zhang, M.-C.; Guo, H.-Y.; Wang, X.-C. Asymmetric C3-Allylation of Pyridines. J. Am. Chem. Soc. 2023, 145, 11789–11797. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.J.; Karakhanov, E.A. Preparation of some N-substituted 1,4-dihydropyridines by metal–ammonia reactions. J. Chem. Soc. Chem. Commun. 1975, 12, 480–481. [Google Scholar] [CrossRef]
- Schlosser, M.; Schneider, P. Metalation of Pyrans and Dihydropyridines: When is an 8 pi-System Cost Effective? Angew. Chem. Int. Ed. 1979, 18, 489–490. [Google Scholar] [CrossRef]
- De Koning, A.J.; Budzelaar, P.H.M.; Brandsma, L.; de Bie, M.J.A.; Boersma, J. Synthesis and NMR spectroscopic properties of some methyl-substituted 1-methyl-1,4-dihydropyridines. Tetrahedron Lett. 1980, 21, 2105–2108. [Google Scholar] [CrossRef]
- Akiyama, K.; Ishii, T.; Tero-Kubota, S.; Ikegami, Y. Photolytic Generation and the Subsequent Dimerization of 4-Alkyl-1-methylpyridinyl Radicals in Solution as Studied by Steady-state and Kinetic ESR Spectroscopy. Bull. Chem. Soc. Jpn. 1985, 58, 3535–3539. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. [Google Scholar] [CrossRef]
- Jupp, A.R.; Stephan, D.W. New Directions for Frustrated Lewis Pair Chemistry. Trends Chem. 2019, 1, 35–48. [Google Scholar] [CrossRef]
- Mahdi, T.; del Castillo, J.N.; Stephan, D.W. Metal-Free Hydrogenation of N-Based Heterocycles. Organometallics 2013, 32, 1971–1978. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Meng, W.; Feng, X.; Yang, J.; Du, H. Borane-Catalyzed Transfer Hydrogenations of Pyridines with Ammonia Borane. Org. Lett. 2016, 18, 5189–5191. [Google Scholar] [CrossRef]
- Eisenberger, P.; Bestvater, B.P.; Keske, E.C.; Crudden, C.M. Hydrogenations at Room Temperature and Atmospheric Pressure with Mesoionic Carbene-Stabilized Borenium Catalysts. Angew. Chem. Int. Ed. 2015, 54, 2467–2471. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Luo, H.; Zhang, M.; Wang, X.-C. Borane-Catalyzed Reduction of Pyridines via a Hydroboration/Hydrogenation Cascade. ACS Catal. 2021, 11, 10824–10829. [Google Scholar] [CrossRef]
- Fan, X.; Zheng, J.; Li, Z.H.; Wang, H. Organoborane Catalyzed Regioselective 1,4-Hydroboration of Pyridines. J. Am. Chem. Soc. 2015, 137, 4916–4919. [Google Scholar] [CrossRef]
- Gandhamsetty, N.; Park, S.; Chang, S. Selective Silylative Reduction of Pyridines Leading to Structurally Diverse Azacyclic Compounds with the Formation of sp3 C–Si Bonds. J. Am. Chem. Soc. 2015, 137, 15176–15184. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Wen, Z.-H.; Wang, X.-C. B(C6F5)3-Catalyzed Cascade Reduction of Pyridines. Angew. Chem. Int. Ed. 2017, 56, 5817–5820. [Google Scholar] [CrossRef]
- Greßies, S.; Süße, L.; Casselman, T.; Stoltz, B.M. Tandem Dearomatization/Enantioselective Allylic Alkylation of Pyridines. J. Am. Chem. Soc. 2023, 145, 11907–11913. [Google Scholar] [CrossRef]
- Bormann, N.; Ward, J.S.; Bergmann, A.K.; Wenz, P.; Rissanen, K.; Gong, Y.; Hatz, W.-B.; Burbaum, A.; Mulks, F.F. Diiminium Nucleophile Adducts are Stable and Convenient Strong Lewis Acids. Chem. Eur. J. 2023, e202302089. [Google Scholar] [CrossRef]
- Greb, L. Lewis Superacids: Classifications, Candidates, and Applications. Chem. Eur. J. 2018, 24, 17881–17896. [Google Scholar] [CrossRef]
- Anders, E.; Opitz, A.; Wermann, K.; Wiedel, B.; Walther, M.; Imhof, W.; Gorls, H. Preparation and Conversion of N-Halomethylpyridinium Halides. Comparison with Related Compounds. J. Org. Chem. 1999, 64, 3113–3121. [Google Scholar] [CrossRef]
- Ho Lee, P.; Lee, K.; Hwan Shim, J.; Guk Lee, S.; Kim, S. Regioselective Synthesis of 4-Alkylpyridines from Pyridine and Aldehydes via Dipole Reversal Process of 1,4-Dihydropyridine Phosphonate. Heterocycles 2006, 67, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Gruzman, D.; Martin, J.M.L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hättig, C.; Höfener, S. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Chem. Phys. 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Chem. Phys. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Böhrer, H.; Trapp, N.; Himmel, D.; Schleep, M.; Krossing, I. From unsuccessful H2-activation with FLPs containing B(Ohfip) 3 to a systematic evaluation of the Lewis acidity of 33 Lewis acids based on fluoride, chloride, hydride and methyl ion affinities. Dalton Trans. 2015, 44, 7489–7499. [Google Scholar] [CrossRef]
- Erdmann, P.; Leitner, J.; Schwarz, J.; Greb, L. An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors. ChemPhysChem 2020, 21, 987–994. [Google Scholar] [CrossRef]
- Erdmann, P.; Greb, L. Multidimensional Lewis Acidity: A Consistent Data Set of Chloride, Hydride, Methide, Water and Ammonia Affinities for 183 p-Block Element Lewis Acids. ChemPhysChem 2021, 22, 935–943. [Google Scholar] [CrossRef]
- Jupp, A.R.; Johnstone, T.C.; Stephan, D.W. The global electrophilicity index as a metric for Lewis acidity. Dalton Trans. 2018, 47, 7029–7035. [Google Scholar] [CrossRef] [PubMed]
- Christe, K.O.; Dixon, D.A.; McLemore, D.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. On a quantitative scale for Lewis acidity and recent progress in polynitrogen chemistry. J. Fluorine Chem. 2000, 101, 151–153. [Google Scholar] [CrossRef]
- Vogler, M.; Süsse, L.; Lafortune, J.H.W.; Stephan, D.W.; Oestreich, M. Electrophilic Phosphonium Cations as Lewis Acid Catalysts in Diels-Alder Reactions and Nazarov Cyclizations. Organometallics 2018, 37, 3303–3313. [Google Scholar] [CrossRef]
- Li, M.L.; Yu, J.H.; Li, Y.H.; Zhu, S.F.; Zhou, Q.L. Highly enantioselective carbene insertion into N-H bonds of aliphatic amines. Science 2019, 366, 990–994. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Mnova; vesion 14.2.3; Mestrelab Research, S.L.: Santiago de Compostela, Spain, 2022.
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- CrysAlisPro; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2018.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Silcock, J.J.; Sterling, A.J.; Duarte, F. autodE: Automated Calculation of Reaction Energy Profiles— Application to Organic and Organometallic Reactions. Angew. Chem. Int. Ed. 2021, 60, 4266–4274. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Born, M. Volumen und Hydratationswärme der Ionen. Z. Phys. 1920, 1, 45–48. [Google Scholar] [CrossRef]
- Klopman, G. Solvations: A semi-empirical procedure for including solvation in quantum mechanical calculations of large molecules. Chem. Phys. Lett. 1967, 1, 200–202. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]
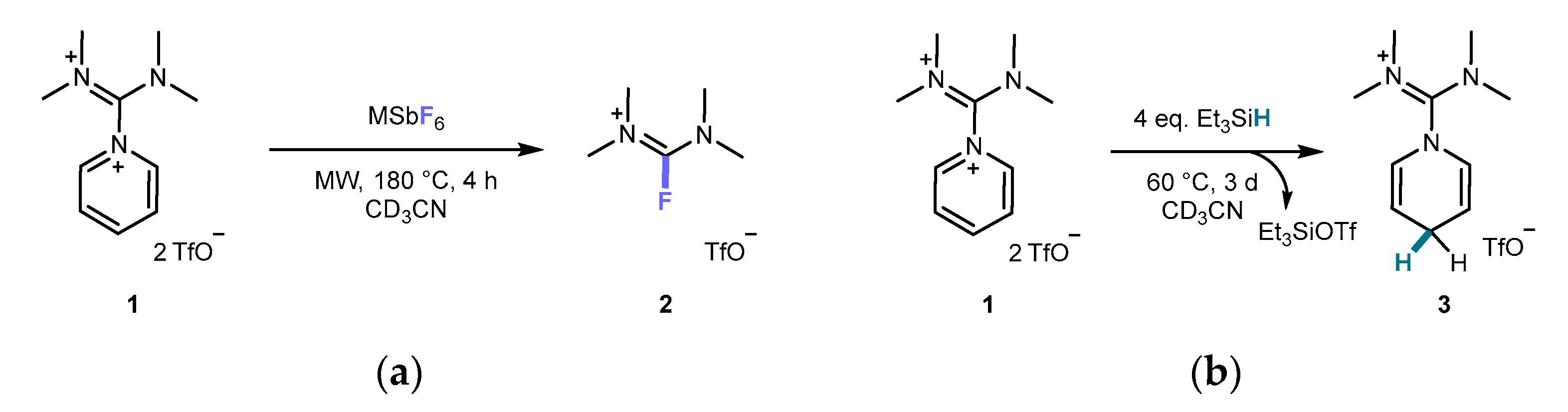
Figure 1.
(a) DIPy 1 abstracts fluoride from various MSbF6 under microwave (MW) conditions in an addition-elimination “double-action” mechanism, yielding fluoroformamidinium 2. (b) The reaction of DIPy 1 with Et3SiH leads to the diiminium-functionalized 1,4-DHP 3.
Figure 1.
(a) DIPy 1 abstracts fluoride from various MSbF6 under microwave (MW) conditions in an addition-elimination “double-action” mechanism, yielding fluoroformamidinium 2. (b) The reaction of DIPy 1 with Et3SiH leads to the diiminium-functionalized 1,4-DHP 3.

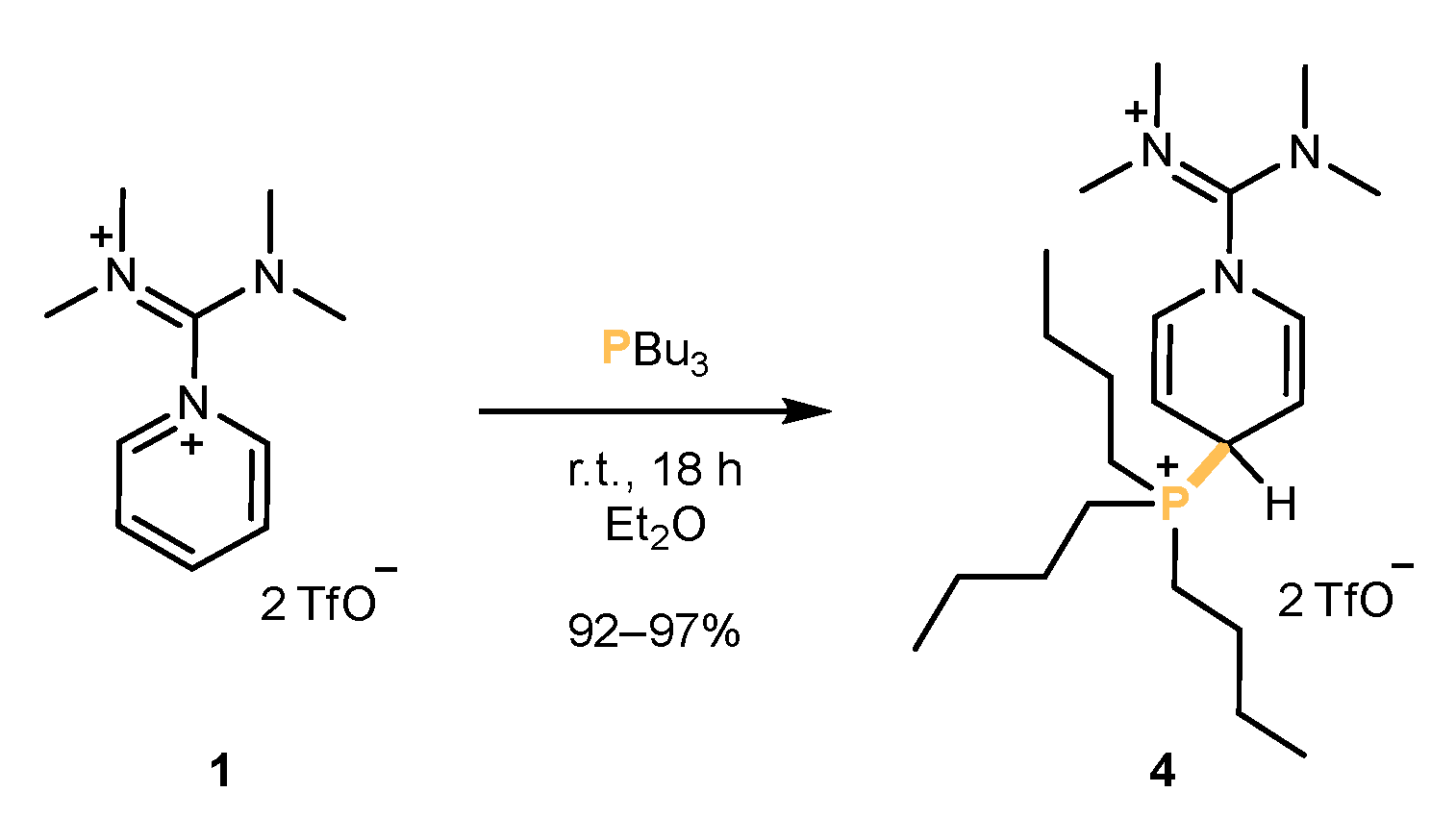
Figure 2.
The synthesis of the phosphonium salt 4 succeeded using a simple addition reaction.

Figure 3.
Structure of 4 determined by SCXRD. Thermal ellipsoids are shown at 50% probability level. Minor disordered position of TfO− omitted and displayed TfO− and butyls simplified for clarity. S: yellow, P: orange, F: green, O: red, N: blue, C: grey, H: white.
Figure 3.
Structure of 4 determined by SCXRD. Thermal ellipsoids are shown at 50% probability level. Minor disordered position of TfO− omitted and displayed TfO− and butyls simplified for clarity. S: yellow, P: orange, F: green, O: red, N: blue, C: grey, H: white.

Figure 4.
1H NMR spectra of 4 (600 MHz, CD3CN). (a) Full spectrum. (b) DHP signals. (c) Diiminium methyl signals of an overlay of 1H NMR spectra taken at 600 and 400 MHz.
Figure 4.
1H NMR spectra of 4 (600 MHz, CD3CN). (a) Full spectrum. (b) DHP signals. (c) Diiminium methyl signals of an overlay of 1H NMR spectra taken at 600 and 400 MHz.

Figure 5.
Computed ion affinities (XIA) in the gas phase and in MeCN solution (given in brackets) in kJ mol−1: (a) FIA, (b) HIA.
Figure 5.
Computed ion affinities (XIA) in the gas phase and in MeCN solution (given in brackets) in kJ mol−1: (a) FIA, (b) HIA.

Figure 6.
(a) Fukui function f+(r) of 4 displayed at ρ = 0.01. (b) Molecular electrostatic potential projected onto the electron density of 4 displayed at ρ = 0.01. Positive: red, negative: blue.
Figure 6.
(a) Fukui function f+(r) of 4 displayed at ρ = 0.01. (b) Molecular electrostatic potential projected onto the electron density of 4 displayed at ρ = 0.01. Positive: red, negative: blue.

Figure 7.
Computed pKa values of 4. a Hoffmann-type elimination of butene was computed.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gong, Y.; Ward, J.S.; Rissanen, K.; Mulks, F.F. Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate. Molbank 2023, 2023, M1710. https://doi.org/10.3390/M1710
AMA Style
Gong Y, Ward JS, Rissanen K, Mulks FF. Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate. Molbank. 2023; 2023(3):M1710. https://doi.org/10.3390/M1710
Chicago/Turabian StyleGong, Yiwei, Jas S. Ward, Kari Rissanen, and Florian F. Mulks. 2023. "Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate" Molbank 2023, no. 3: M1710. https://doi.org/10.3390/M1710
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.