
Are Therapies That Target α-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review

Abstract
:1. Introduction
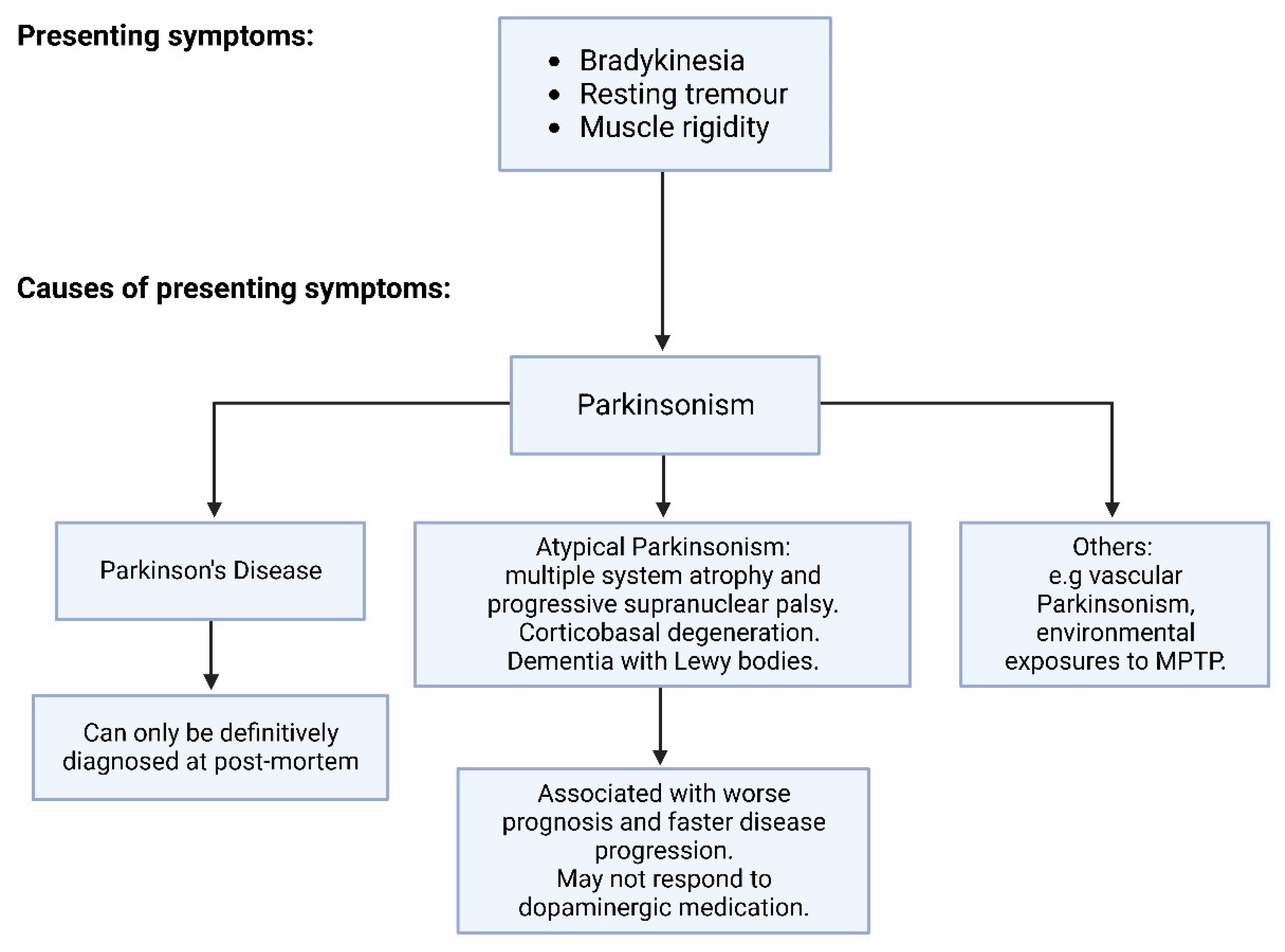
1.1. Pathophysiology of Parkinson’s Disease
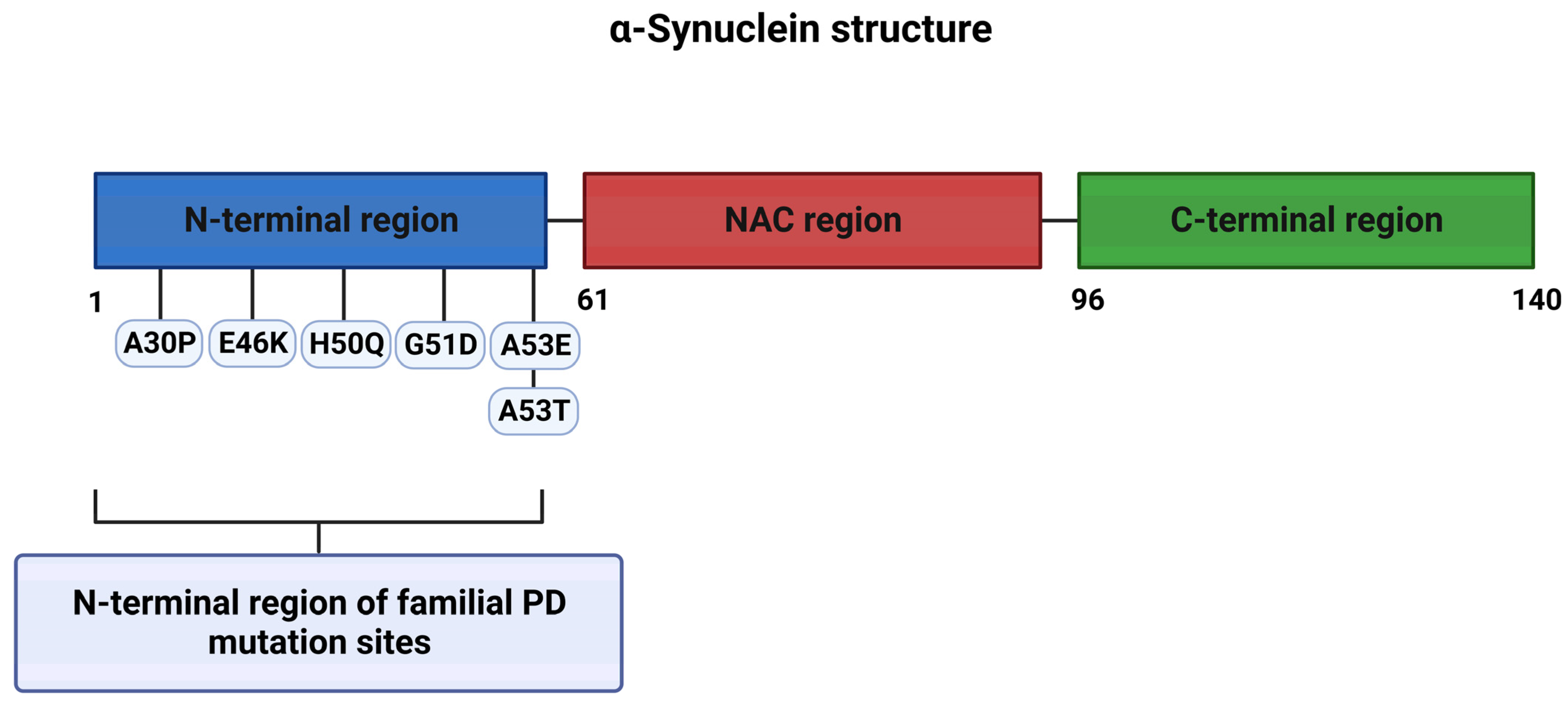
1.1.1. Genetics of PD
1.1.2. Neuroanatomy of PD
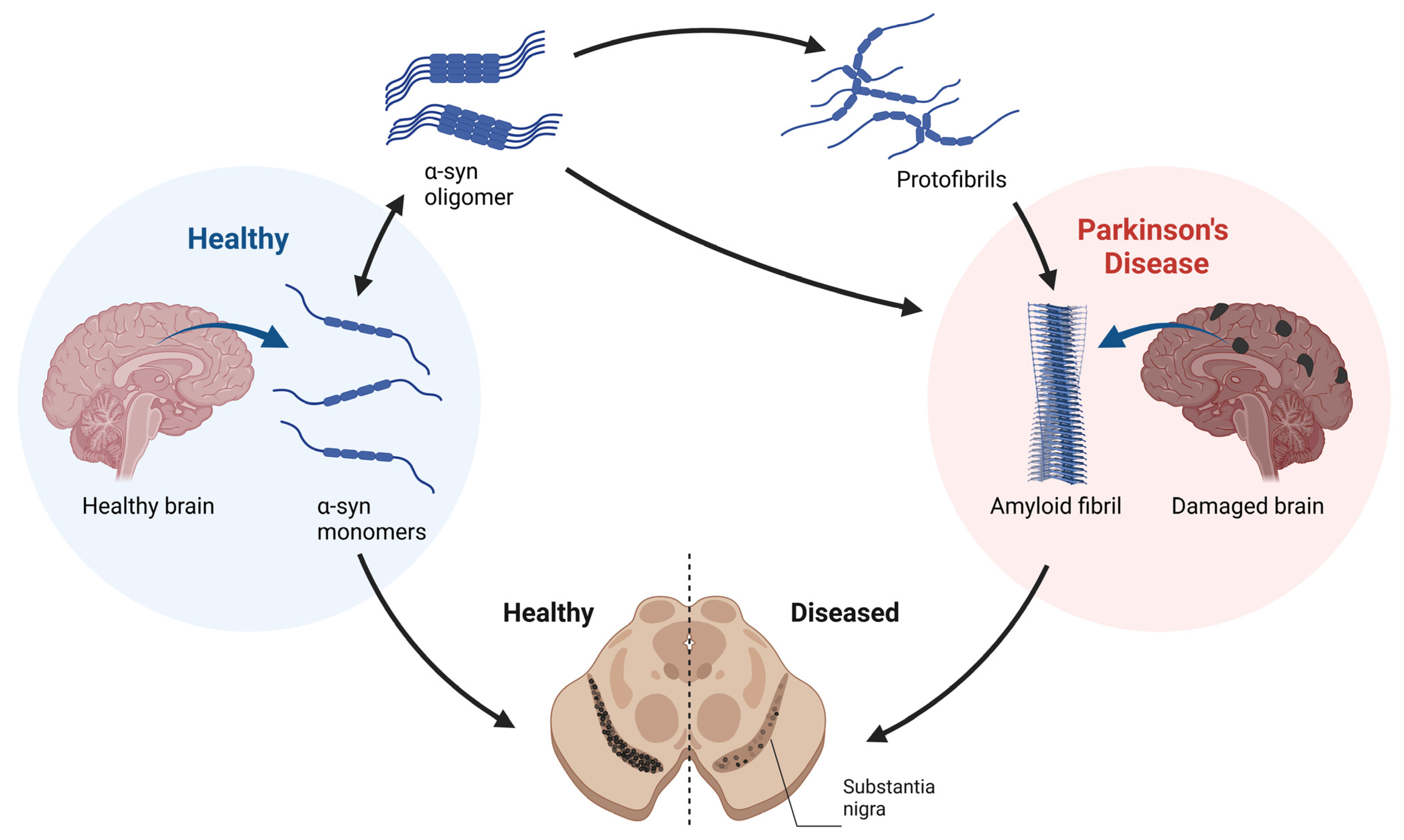
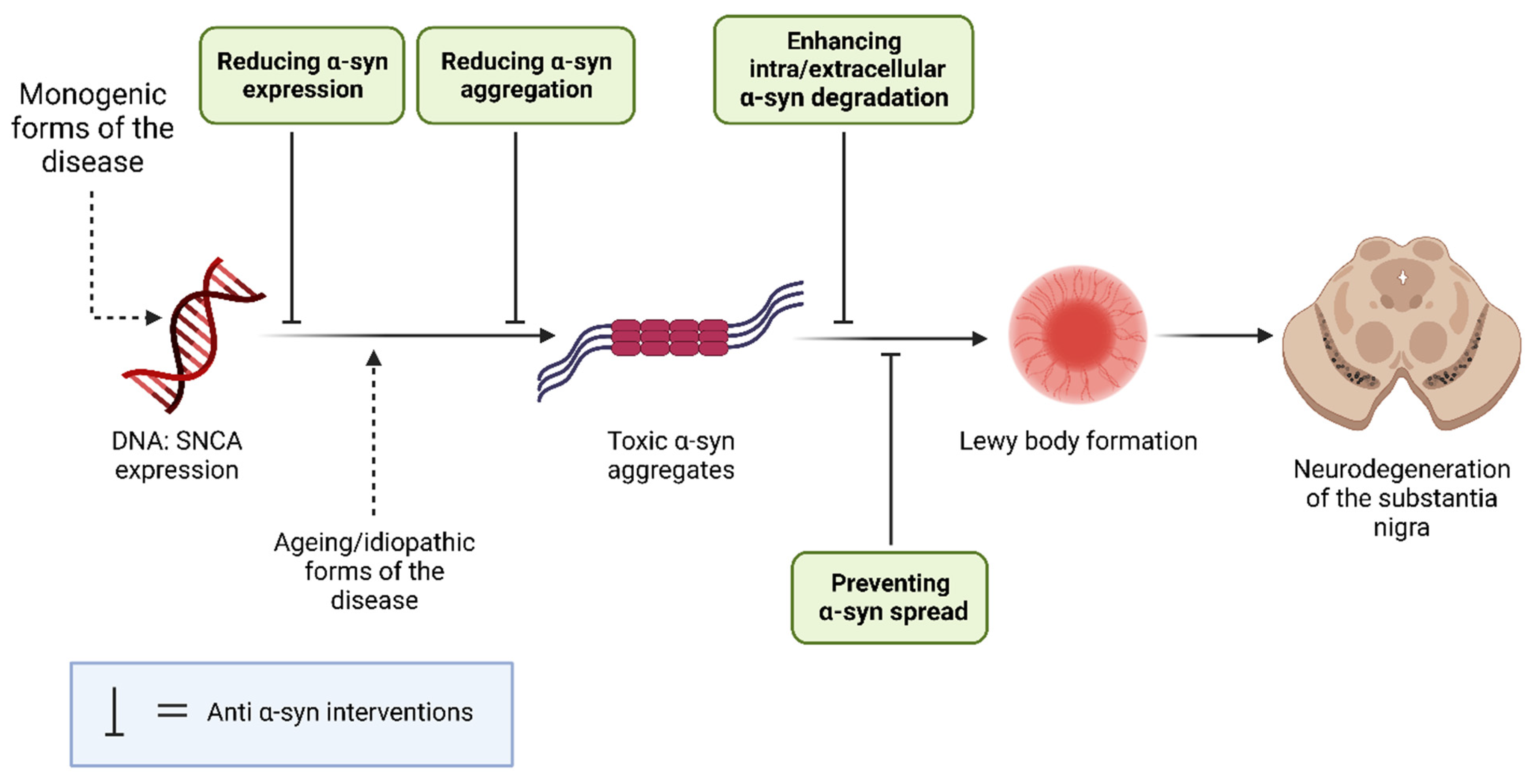
1.2. α-Syn Toxicity in PD
1.3. α-Synuclein as a PD Therapeutic Target
2. Materials and Methods
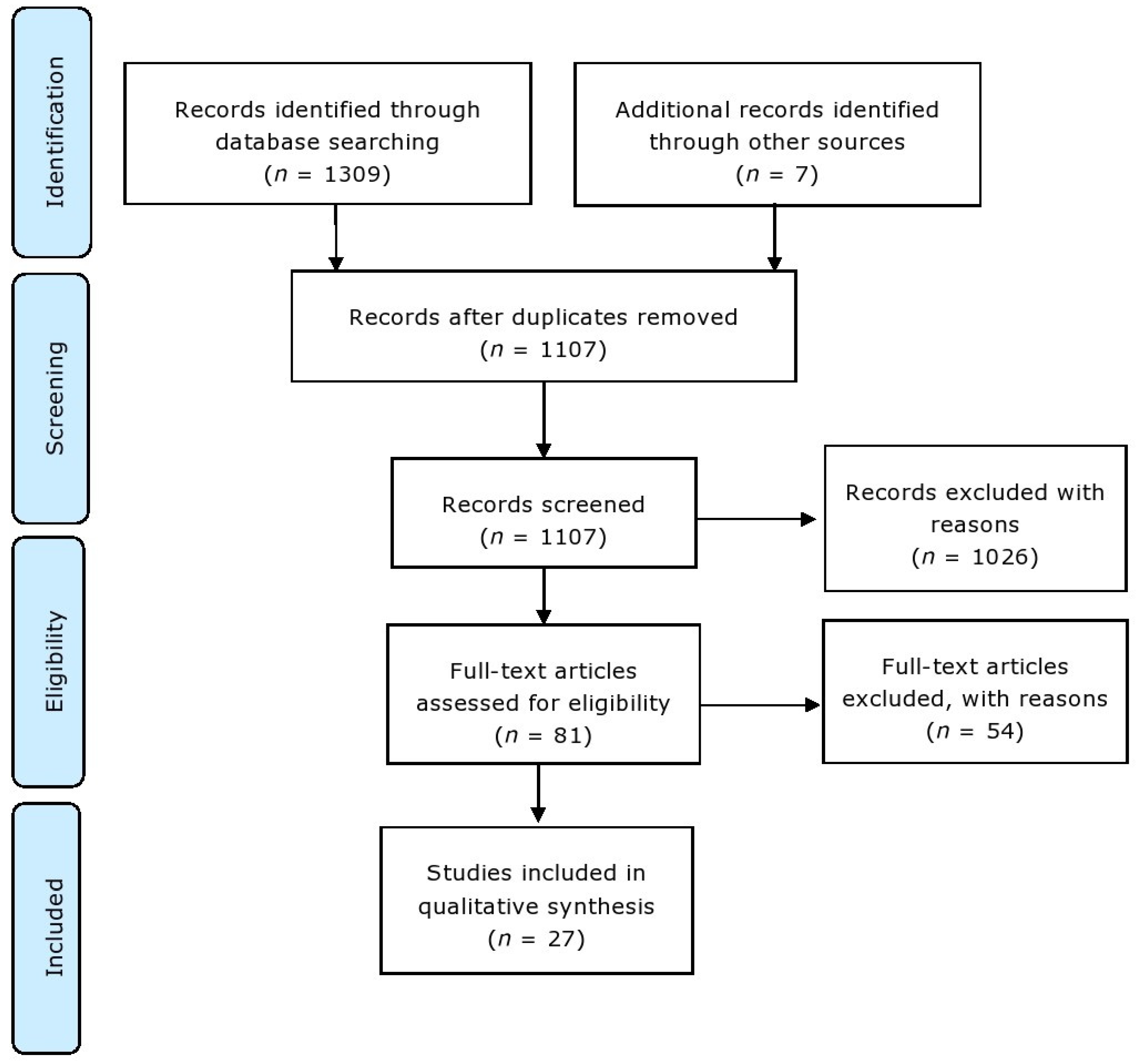
2.1. Information Sources and Search Strategy
2.2. Eligibility Criteria
2.3. Study Selection and Data Collection Process
3. Results
3.1. Background Mechanisms
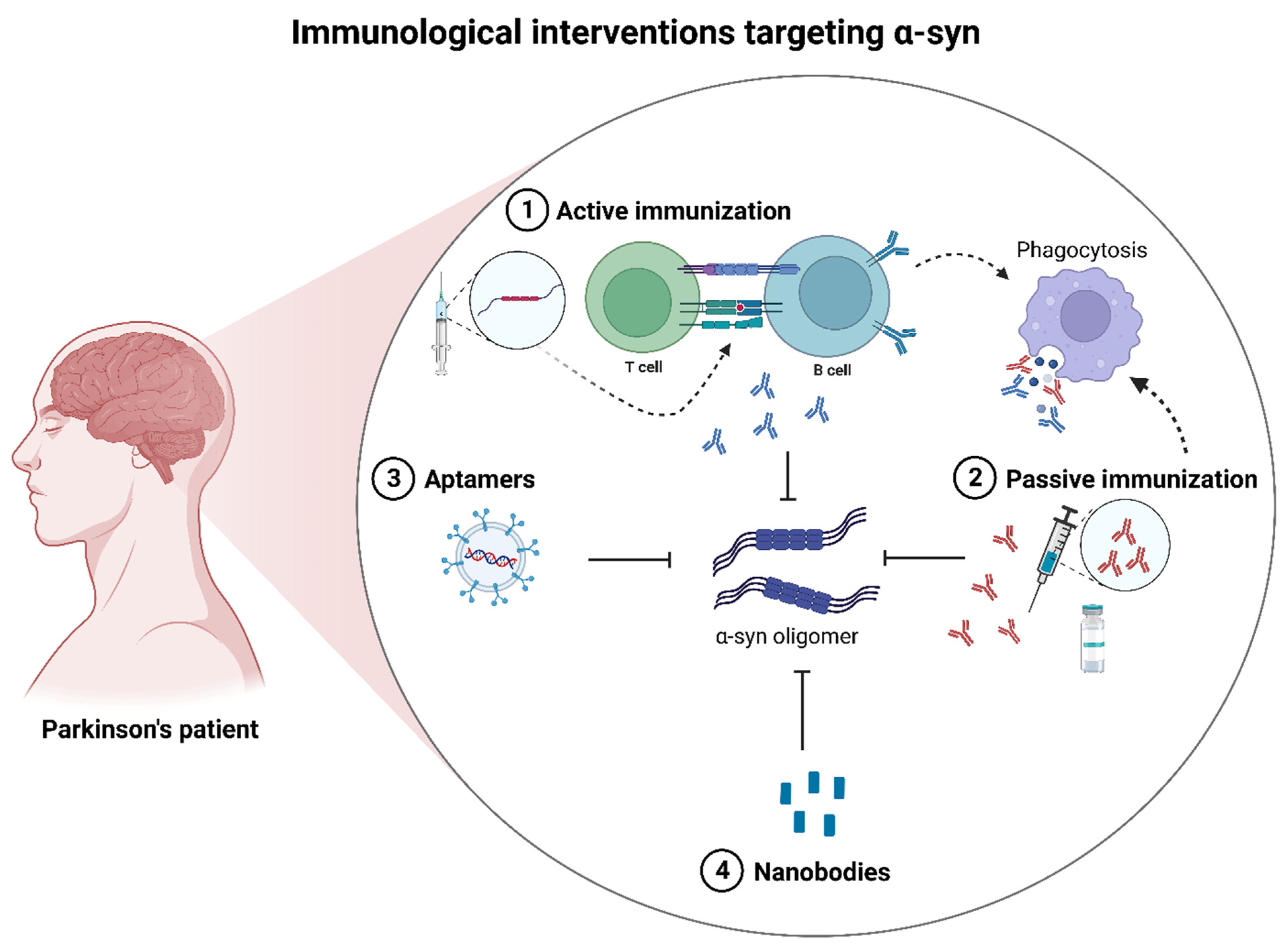
3.1.1. Immunotherapeutic Interventions
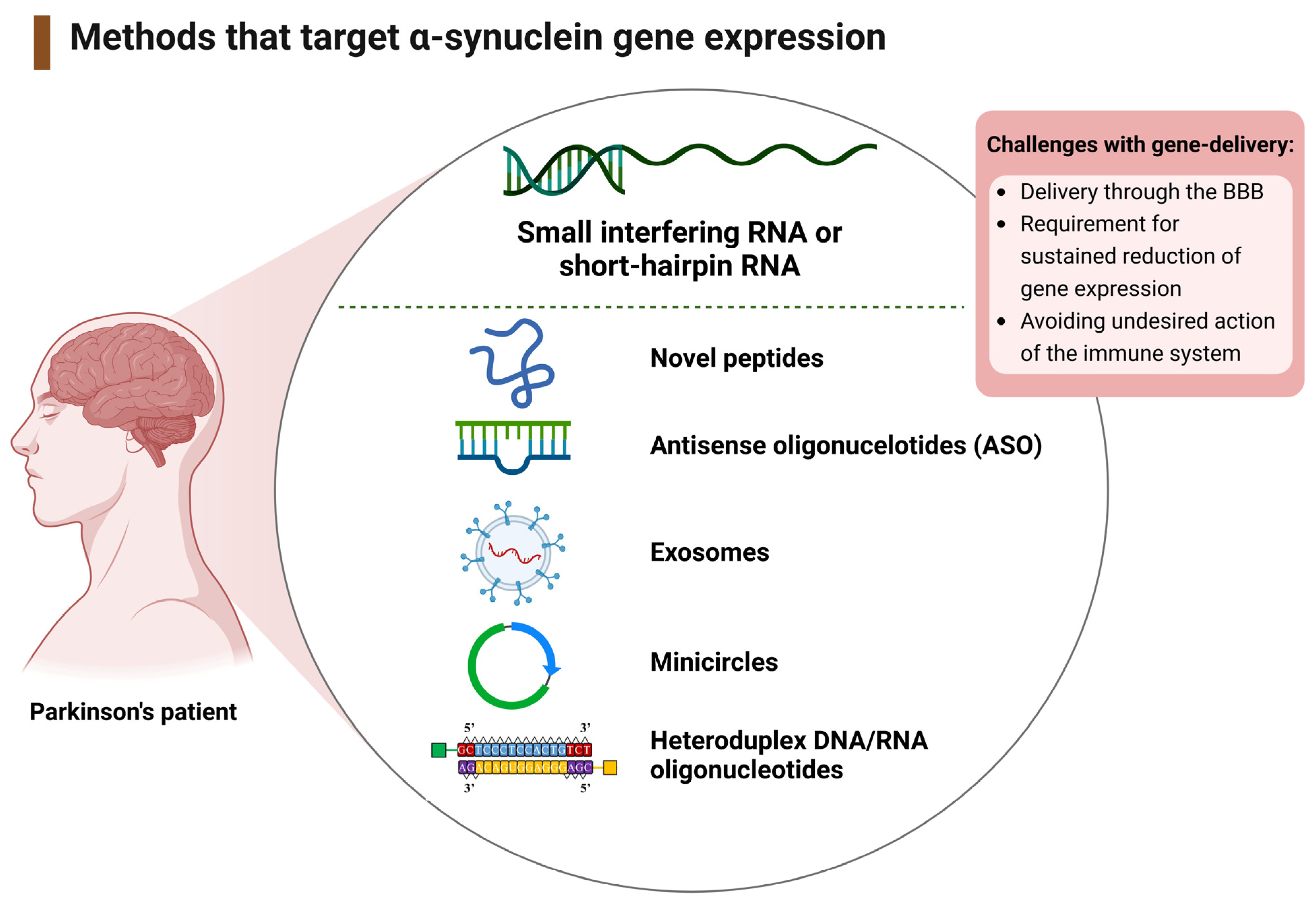
3.1.2. Therapeutic Interventions That Target the SNCA Gene
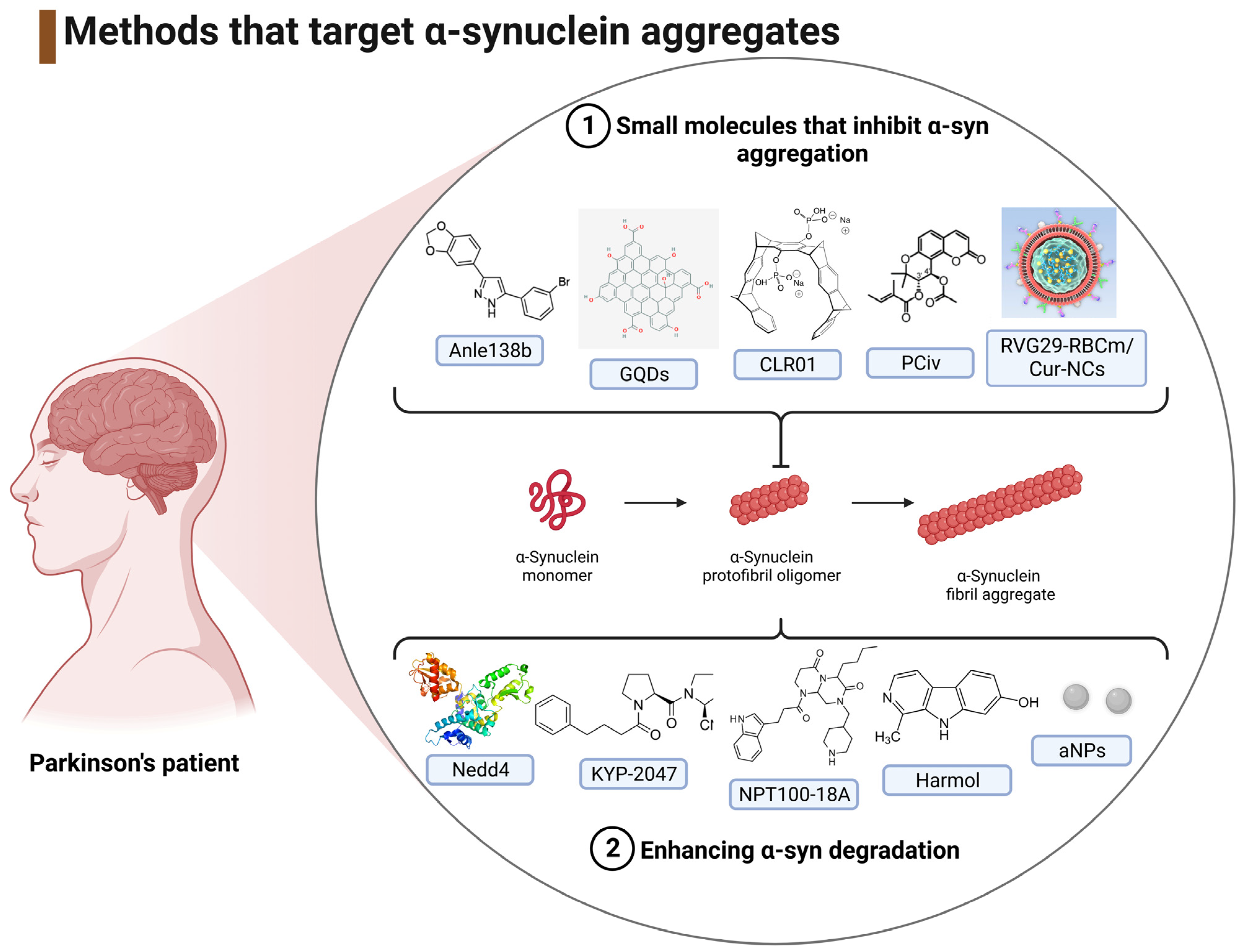
3.1.3. Interventions That Target the Reduction of α-Syn Aggregates
3.2. Study Characteristics
3.3. Biochemical and Immunohistochemical Outcomes
3.3.1. Immunotherapeutic Outcomes
3.3.2. Gene-Therapy Outcomes
3.3.3. Outcomes from Agents That Reduce the Levels of α-Syn Aggregates
4. Discussion
4.1. Mechanisms of Immunotherapeutic Interventions Targeting α-Syn
4.1.1. Active Immunotherapy
4.1.2. Passive Immunotherapy
4.1.3. Alternative Immunotherapies
4.1.4. Immunotherapeutic Conclusions
4.2. Mechanisms of Targeting SNCA Expression
Viral and Non-Viral Delivery of RNA-Based Gene Therapy
4.3. Inhibition of α-Syn Aggregation
4.3.1. Small Molecules That Inhibit α-Syn Aggregation
4.3.2. Enhancing α-Syn Degradation
4.4. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- United Nations. Department of Economic and Social Affairs. World Population Prospects. World Population Prospects. 2019. Available online: https://population.un.org/wpp/Publications/Files/WPP2019_Highlights.pdf (accessed on 28 June 2023).
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M.; Klein, C. Epidemiology and causes of Parkinson’s disease. Nervenarzt 2017, 88, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2018, 16, 1239–1252. [Google Scholar] [CrossRef]
- Domingo, A.; Klein, C. Genetics of Parkinson Disease. In The Handbook of Clinical Neurology; North-Holland Publishing Company: Amsterdam, The Netherlands, 2018; Volume 147, Chapter 14; pp. 211–227. [Google Scholar] [CrossRef]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell. Probes 2016, 30, 386–396. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Post, M.R.; Lieberman, O.J.; Mosharov, E.V. Can Interactions Between α-synuclein, Dopamine and Calcium Explain Selective Neurodegeneration in Parkinson’s Disease? Front. Neurodegener. 2018, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Di Lazzaro, G.; Marino, G.; Campanelli, F.; Ghiglieri, V. Advances in understanding the function of alpha-synuclein: Implications for Parkinson’s disease. Brain 2023, awad150. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneswara, V.; Cass, S.; Wayne, D.; Bolt, E.L.; Ray, D.E.; Carter, W.G. Molecular ageing of alpha- and Betasynucleins: Protein damage and repair mechanisms. PLoS ONE 2013, 8, e61442. [Google Scholar] [CrossRef] [Green Version]
- Schaffert, L.N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-syn oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef] [Green Version]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; More, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.; Rüb, U.; De Vos, R.; Jansen Steur, E.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Ageing 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kwon, S.; Kam, T.; Panicker, N.; Karuppagounder, S.; Lee, S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neurone 2019, 103, 627–641. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.; Kordower, J. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.; Bengoa-vergniory, N.; Wade-martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.J.; Mckenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Sue, E. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. PLoS Med. 2021, 18, 1003583. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, D.; Kordower, J. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104587. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Evaluating the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of BIIB054 in Participants with Parkinson’s Disease (SPARK). 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT03318523 (accessed on 10 May 2023).
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of UB-312 in Healthy Participants and Parkinson’s Disease Patients. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04075318?id=NCT01885494+OR+NCT01568099+OR+NCT02216188+OR+NCT02914366+OR+NCT04075318+OR+NCT04127695&draw=2&rank=2&load=cart (accessed on 10 May 2023).
- Sanchez-Guajardo, V.; Annibali, A.; Jensen, P.H.; Romero-Ramos, M. Alpha-Synuclein Vaccination Prevents the Accumulation of Parkinson Disease-Like Pathologic Inclusions in Striatum in Association with Regulatory T Cell Recruitment in a Rat Model. J. Neuropathol. Exp. Neurol. 2013, 72, 624–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an alpha-synuclein-based Parkinson’s disease model. Npj Park. Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.; Schenk, D.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-α-Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Study to Evaluate the Efficacy of Prasinezumab (RO7046015/PRX002) in Participants with Early Parkinson’s Disease (PASADENA). 2023. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03100149 (accessed on 10 May 2023).
- Ren, X.X.; Zhao, Y.; Xue, F.Q.; Zheng, Y.; Huang, H.X.; Wang, W.; Chang, Y.; Yang, H.; Zhang, J. Exosomal DNA Aptamer Targeting alpha-Synuclein Aggregates Reduced Neuropathological Deficits in a Mouse Parkinson’s Disease Model. Mol. Ther. Nucleic Acids 2019, 17, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef] [PubMed]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Volc, D.; Seppi, K.; Medori, R.; Lührs, P.; Kutzelnigg, A.; Djamshidian, A.; Thun-Hohenstein, C.; Meissner, W.G.; Rascol, O.; et al. Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study. J. Park. Dis. 2021, 11, 1079–1089. [Google Scholar] [CrossRef]
- Butler, Y.R.; Liu, Y.; Kumbhar, R.; Zhao, P.; Gadhave, K.; Wang, N.; Li, Y.; Mao, X.; Wang, W. α-Synuclein fibril-specific nanobody reduces prion-like α-synuclein spreading in mice. Nat. Commun. 2022, 13, 4060. [Google Scholar] [CrossRef]
- Schmidhuber, S.; Scheiblhofer, S.; Weiss, R.; Cserepes, M.; Tovari, J.; Gadermaier, G.; Bezard, E.; De Giorgi, F.; Ichas, F.; Strunk, D.; et al. A Novel C-Type Lectin Receptor-Targeted alpha-Synuclein-Based Parkinson Vaccine Induces Potent Immune Responses and Therapeutic Efficacy in Mice. Vaccines 2022, 10, 1432. [Google Scholar] [CrossRef]
- Roshanbin, S.; Julku, U.; Xiong, M.F.; Eriksson, J.; Masliah, E.; Hultqvist, G.; Bergstrom, J.; Ingelsson, M.; Syvanen, S.; Sehlin, D. Reduction of alpha SYN Pathology in a Mouse Model of PD Using a Brain-Penetrating Bispecific Antibody. Pharmaceutics 2022, 14, 1412. [Google Scholar] [CrossRef]
- Helmschrodt, C.; Hobel, S.; Schoniger, S.; Bauer, A.; Bonicelli, J.; Gringmuth, M.; Fietz, S.A.; Aigner, A.; Richter, A.; Richter, F. Polyethylenimine Nanoparticle-Mediated siRNA Delivery to Reduce alpha-Synuclein Expression in a Model of Parkinson’s Disease. Mol. Ther.—Nucelic Acids 2017, 9, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izco, M.; Blesa, J.; Schleef, M.; Schmeer, M.; Porcari, R.; Al-Shawi, R.; Ellmerich, S.; de Toro, M.; Gardiner, C.; Seow, Y.; et al. Systemic Exosomal Delivery of shRNA Minicircles Prevents Parkinsonian Pathology. Mol. Ther. 2019, 27, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; El-Agnaf, O.M.A.; Kim, C.; Masliah, E.; Rissman, R.A. Systemic peptide mediated delivery of an siRNA targeting alpha-syn in the CNS ameliorates the neurodegenerative process in a transgenic model of Lewy body disease. Neurobiol. Dis. 2019, 127, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.; Zhao, H.; Collier, T.; Sandoval, I.; Sortwell, C.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Fan, X.; Del Cid-Pellitero, E.; Liu, X.; Zhou, L.; Dai, C.; Gibbs, E.; He, W.; Li, H.; Wu, X.; et al. Development of an alpha-synuclein knockdown peptide and evaluation of its efficacy in Parkinson’s disease models. Commun. Biol. 2021, 4, 232. [Google Scholar] [CrossRef]
- Cao, Q.; Luo, S.; Yao, W.; Qu, Y.; Wang, N.; Hong, J.; Murayama, S.; Zhang, Z.; Chen, J.; Hashimoto, K.; et al. Suppression of abnormal alpha-synuclein expression by activation of BDNF transcription ameliorates Parkinson’s disease-like pathology. Mol. Ther.-Nucleic Acids 2022, 29, 1–15. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.; Hallett, P.; Moens, T.; Smith, G.; Mangano, E.; Kim, H.; Goldberg, A.L.; Liu, J.-L.; Isacson, O.; Tofaris, G.K. Enhanced ubiquitin-dependent degradation by Nedd4 protects against alpha-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 2014, 64, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Mannisto, P.T.; Maguire-Zeiss, K.A.; Myohanen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain J. Neurol. 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Yoo, J.M.; Hwang, H.; Lee, J.; Lee, S.H.; Yun, S.P.; Park, M.J.; Lee, M.; Choi, S.; Kwon, S.H.; et al. Graphene quantum dots prevent alpha-synucleinopathy in Parkinson’s disease. Nat. Nanotechnol. 2018, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Bengoa-Vergniory, N.; Faggiani, E.; Ramos-Gonzalez, P.; Kirkiz, E.; Connor-Robson, N.; Brown, L.V.; Siddique, I.; Li, Z.; Vingill, S.; Cioroch, M.; et al. CLR01 protects dopaminergic neurons in vitro and in mouse models of Parkinson’s disease. Nat. Commun. 2020, 11, 4885. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ao, Y.L.; Huang, C.H.; Song, X.B.; Zhang, G.L.; Cui, W.; Wang, Y.Q.; Zhang, X.Q.; Zhang, Z.J. Harmol promotes alpha-synuclein degradation and improves motor impairment in Parkinson’s models via regulating autophagy-lysosome pathway. Npj Park. Dis. 2022, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.L.; Soria, F.N.; Cunha, A.; Doudnikoff, E.; Prévot, G.; Daniel, J.; Blanchard-Desce, M.; Barthélémy, P.; Bezard, E.; Crauste-Manciet, S.; et al. Acidic nanoparticles protect against α-synuclein-induced neurodegeneration through the restoration of lysosomal function. Aging Cell 2022, 21, e13584. [Google Scholar] [CrossRef]
- Kim, H.; Maeng, H.-J.; Kim, J.H.; Yoon, J.-H.; Oh, Y.; Paek, S.-M.; Lee, Y. Synthetic Peucedanocoumarin IV Prevents α-Synuclein Neurotoxicity in an Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 8618. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, J.; Liu, Y.; Liu, W.; Yu, G.; Huang, Y.; Yang, Y.; Chen, X.; Chen, T. Brain-Targeted Biomimetic Nanodecoys with Neuroprotective Effects for Precise Therapy of Parkinson’s Disease. ACS Cent. Sci. 2022, 8, 1336–1349. [Google Scholar] [CrossRef]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine Hydroxylase and Regulation of Dopamine Synthesis. Arch. Biochem. Biophys. 2010, 508, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Baecher-Allan, C.; Kaskow, B.; Weiner, H. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Nimmo, J.T.; Smith, H.; Wang, C.Y.; Teeling, J.L.; Nicoll, J.; Verma, A.; Dodart, J.-C.; Liu, Z.; Lin, F.; Carare, R.O. Immunisation with UB-312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α-synuclein accumulation in the brain and gut. Acta Neuropathol. 2020, 143, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Hovakimyan, A.; Zagorski, K.; Antonyan, T.; Petrushina, I.; Davtyan, H.; Chailyan, G.; Hasselmann, J.; Iba, M.; Adame, A.; et al. Efficacy and immunogenicity of MultiTEP-based DNA vaccines targeting human α-synuclein: Prelude for IND enabling studies. Npj Vaccines 2022, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Zagorski, K.; Chailyan, G.; Hovakimyan, A.; Antonyan, T.; Kiani Shabestari, S.; Petrushina, I.; Davtyan, H.; Cribbs, D.H.; Blurton-Jones, M.; Masliah, E.; et al. Immunogenicity of MultiTEP-Platform-Based Recombinant Protein Vaccine, PV-1950R, Targeting Three B-Cell Antigenic Determinants of Pathological α-Synuclein. Int. J. Mol. Sci. 2022, 23, 6080. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Valera, E.; Rockenstein, E.; Overk, C.; Mante, M.; Adame, A.; Zago, W.; Seubert, P.; Barbour, R.; Schenk, D.; et al. Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol. Commun. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Nordström, E.; Eriksson, F.; Sigvardson, J.; Johannesson, M.; Kasrayan, A.; Jones-Kostalla, M.; Appelkvist, P.; Söderberg, L.; Nygren, P.; Blom, M.; et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study to Evaluate the Safety and Tolerability of ABBV-0805 in Patients with Parkinson’s Disease. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04127695 (accessed on 10 May 2023).
- Jin, B.-K.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int. J. Mol. Sci. 2023, 24, 5994. [Google Scholar] [CrossRef]
- Fassler, M.; Benaim, C.; George, J. A Single Chain Fragment Variant Binding Misfolded Alpha-Synuclein Exhibits Neuroprotective and Antigen-Specific Anti-Inflammatory Properties. Cells 2022, 11, 3822. [Google Scholar] [CrossRef]
- Ni, S.; Zhuo, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y.; et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces 2021, 13, 9500–9519. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood J. Am. Soc. Hematol. 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Sabin, J.; Alatorre-Meda, M.; Miñones JJr Domínguez-Arca, V.; Prieto, G. New insights on the mechanism of polyethylenimine transfection and their implications on gene therapy and DNA vaccines. Colloids Surf. B Biointerfaces 2022, 210, 112219. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S.; et al. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: A randomised, double-blind, placebo-controlled phase 1a trial. EBioMedicine 2022, 80, 104021. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A First-in-Human Study of Single and Multiple Doses of anle138b in Healthy Subjects. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04208152 (accessed on 10 May 2023).
- Kumar, A.; Chaudhary, R.K.; Singh, R.; Singh, S.P.; Wang, S.Y.; Hoe, Z.Y.; Pan, C.-T.; Shiue, Y.-L.; Wei, D.-Q.; Kaushik, A.C.; et al. Nanotheranostic applications for detection and targeting neurodegenerative diseases. Front. Neurosci. 2020, 14, 305. [Google Scholar] [CrossRef] [PubMed]
- Schrader, T.; Bitan, G.; Klärner, F.G. Molecular tweezers for lysine and arginine—Powerful inhibitors of pathologic protein aggregation. Chem. Commun. 2016, 52, 11318–11334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.A.; Kinsman, G.; Kramer, E.R. The Role of NEDD4 E3 Ubiquitin–Protein Ligases in Parkinson’s Disease. Genes 2022, 13, 513. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M. Interaction of prolyl oligopeptidase with α-synuclein. CNS Neurol. Disord. Drug Targets 2011, 10, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Eteläinen, T.S.; Kilpeläinen, T.P.; Ignatius, A.; Auno, S.; De Lorenzo, F.; Uhari-Väänänen, J.K.; Julku, U.H.; Myöhänen, T.T. Removal of proteinase K resistant αSyn species does not correlate with cell survival in a virus vector-based Parkinson’s disease mouse model. Neuropharmacology 2022, 218, 109213. [Google Scholar] [CrossRef]
- Xu, N.; LaGrow, T.J.; Anumba, N.; Lee, A.; Zhang, X.; Yousefi, B.; Bassil, Y.; Clavijo, G.P.; Khalilzad Sharghi, V.; Maltbie, E.; et al. Functional Connectivity of the Brain Across Rodents and Humans. Front. Neurosci. 2022, 16, 816331. [Google Scholar] [CrossRef]
- Beauchamp, A.; Yee, Y.; Darwin, B.C.; Raznahan, A.; Mars, R.B.; Lerch, J.P. Whole-brain comparison of rodent and human brains using spatial transcriptomics. Elife 2022, 11, e79418. [Google Scholar] [CrossRef]
- Prakash, S.; Carter, W.G. The Neuroprotective Effects of Cannabis-Derived Phytocannabinoids and Resveratrol in Parkinson’s Disease: A Systematic Literature Review of Pre-Clinical Studies. Brain Sci. 2021, 11, 1573. [Google Scholar] [CrossRef]
- Ghanem, S.S.; Majbour, N.K.; Vaikath, N.N.; Ardah, M.T.; Erskine, D.; Jensen, N.M.; Fayyad, M.; Sudhakaran, I.P.; Vasili, E.; Melachroinou, K.; et al. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2109617119. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Ikuno, M.; Yamakado, H.; Takahashi, R. Animal Model for Prodromal Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stott, S.R.W.; Wyse, R.K.; Brundin, P. Drug Repurposing for Parkinson’s Disease: The International Linked Clinical Trials experience. Front. Neurosci. 2021, 15, 653377. [Google Scholar] [CrossRef] [PubMed]
- Latif, K.; Ullah, A.; Shkodina, A.D.; Boiko, D.I.; Rafique, Z.; Alghamdi, B.S.; Alfaleh, M.A.; Ashraf, G.M. Drug reprofiling history and potential therapies against Parkinson’s disease. Front. Pharmacol. 2022, 13, 1028356. [Google Scholar] [CrossRef]
- Peña-Díaz, S.; García-Pardo, J.; Ventura, S. Development of Small Molecules Targeting α-Synuclein Aggregation: A Promising Strategy to Treat Parkinson’s Disease. Pharmaceutics 2023, 15, 839. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Agent | Type of Study | Treatment Strategy and Study Objectives |
|---|---|---|---|
| Sanchez-Guajardo et al., 2013 [35] | rAAv-α-syn | in vivo | Application of a neuroprotective vaccine to potentiate natural immune tolerance to α-syn. |
| Chatterjee et al., 2018 [36] | VH14*PEST and NbSyn87*PEST | in vivo | Utilization of human anti-α-syn nanobody constructs fused to a proteasome-targeting PEST sequence to enhance the clearance of target (α-syn) antigens. |
| Jankovic et al., 2018 (NCT02157714) and NCT03100149 [37,38] | PRX002 | Phase I and II Clinical Trials | Assessment of the efficacy and safety of the humanized monoclonal antibody, prasinezumab, directed against the C-terminus of α-syn designed to prevent α-syn aggregation and its cell-to-cell transmission. |
| Ren et al., 2019 [39] | RVG-exosome aptamer | in vivo | RVG-exosome delivery of aptamers that recognize α-syn to reduce the formation of α-syn aggregates. |
| Schofield et al., 2019 [40] | MEDI1341 | in vivo | Application of a high affinity antibody directed to the C-terminal of α-syn to sequester extracellular α-syn and attenuate α-syn spreading. |
| Volc et al., 2020 [41] | PD01A | Phase I Clinical Trial | Assessment of safety and tolerability of epitope mimetics of a C-terminal region of α-syn conjugated to a carrier protein to break immune tolerance and produce antigen-specific antibodies. |
| Poewe et al., 2021 [42] | PD03A | Phase I Clinical Trial | |
| Butler et al., 2022 [43] | AAV-EGFP-PFFNB2 | in vivo | Utilization of an anti-α-syn nanobody (PFFNB2) fused with an AAV-EGFP to dissociate α-syn fibrils and limit α-syn spread. |
| Schmidhuber et al., 2022 [44] | WISIT vaccine | in vivo | Utilization of a DNA vaccine for multiple epitopes to reduce α-syn aggregation and propagation. |
| Roshanbin et al., 2022 [45] | RmAbSynO2-scFv8D3 | in vivo | Utilization of a bispecific antibody to α-syn and the transferrin receptor to facilitate uptake across the BBB to target α-syn aggregates. |
| Study | Agent | Category of Gene Therapy | Treatment Strategy |
|---|---|---|---|
| Helmschrodt et al., 2017 [46] | PEI/SNCA-siRNA | Non-viral | Utilization of nanoparticle PEI to mediate the delivery of siRNA into the brains of mice to reduce expression of the SNCA gene and α-syn protein production. |
| Alarcón-Arís et al., 2018 [47] | IND-siRNA or IND-1233-ASO | Non-viral ASOs | Utilization of indatraline-conjugated ASO or siRNA to knockdown expression of the SNCA gene and α-syn protein production. |
| Izco et al., 2019 [48] | RVG-exosomes with anti-GFP shRNA-MCs | Viral delivery | Nanoparticle delivery of shRNA-MCs into the brain via RVG-exosomes to knockdown SNCA gene expression to limit the formation of α-syn aggregates. |
| Spencer et al., 2019 [49] | ApoB11 | Non-viral | Conjugation of an 11 amino acid sequence of ApoB protein coupled to a 9 amino acid linker to deliver siRNA across the BBB to reduce α-syn levels. |
| Cole et al., 2021 [50] | ASO1 | Non-viral ASOs | Utilization of ASOs targeting the SNCA gene to reduce the production of α-syn protein. |
| Jin et al., 2021 [51] | Tat-βsyn-degron | Non-viral | Utilization of Tat-βsyn-degron, a three-domain synthetic peptide designed to cross the BBB, bind to endogenous α-syn, and target it for proteasomal degradation. |
| Cao et al., 2022 [52] | HDOs | Non-viral | Utilization of HDOs to knockdown expression of the SNCA gene and α-syn protein production. |
| Study | Agent | Intervention Category | Mechanism of Action |
|---|---|---|---|
| Wagner et al., 2013 [53] | Anle138b | Small molecule | Utilization of anle138b as an α-syn aggregation inhibitor derived from DPP. |
| Davies et al., 2014 [54] | Nedd4 | Degradation enhancer | Utilization of Nedd4 as a ubiquitin ligase to target α-syn for lysosomal degradation. |
| Savolainen et al., 2014 [55] | KYP-2047 | Degradation enhancer | Utilization of KYP-2047, a PREP inhibitor, to enhance clearance of α-syn via autophagy. |
| Wrasidlo et al., 2016 [56] | NPT100-18A | Small molecule | Utilization of NPT100-18A, a cyclic peptidomimetic derived from small peptides analogous to the 96–102 domain of α-syn, capable of displacing membrane-associated α-syn and reducing oligomer formation. |
| Kim et al., 2018 [57] | GQDs | Small molecule | Utilization of GQDs to inhibit fibrillization and enhance α-syn disaggregation. |
| Bengoa-Vergniory et al., 2020 [58] | CLR01 | Small molecule | Utilization of CLR01 as a molecular tweezer to decrease α-syn aggregation. |
| Xu et al., 2022 [59] | Harmol | Degradation enhancer | Utilization of harmol to promote α-syn degradation by an autophagic-lysosomal pathway. |
| Arotcarena et al., 2022 [60] | aNPs | Degradation enhancer | Utilization of aNPs to promote α-syn degradation by enhanced lysosomal activity. |
| Kim et al., 2022 [61] | PCiv | Small molecule | Utilization of PCiv as an α-syn disaggregation agent. |
| Liu et al., 2022 [62] | RVG29-RBCm/Cur-NCs | Small molecule | Utilization of RVG29-RBCm/Cur-NCs a nanodecoy to act as an α-syn aggregation inhibitor able to cross the BBB. |
| Study | Agent | Changes in α-Syn and TH in Intervention Groups (vs. PD Model) | Type of α-Syn Species Targeted | Level of Significance |
|---|---|---|---|---|
| Sanchez-Guajardo et al., 2013 [35] | rAAv-α-syn | ↓α-syn aggregates TH levels similar to control | Aggregates | (p < 0.05) ND |
| Chatterjee et al., 2018 [36] | VH14/ NbSyn87*PEST | ↓α-syn aggregates ↑TH-labelled cells | Aggregates | (p < 0.05) (p < 0.01) (VH14*PEST) |
| Jankovic et al., 2018 [37] | PRX002 | ↓Free-to-total serum α-syn | Aggregates | (p < 0.001) |
| Ren et al., 2019 [39] | RVG-exosome aptamer | ↓α-syn | Fibrils aggregates | (p < 0.01) |
| Schofield et al., 2019 [40] | MEDI1341 | ↓α-syn | Oligomers | Hippocampal (p < 0.001) Neocortical (p < 0.001) |
| Volc et al., 2020 [41] | PD01A | ↓α-syn | Oligomers | CSF (↓51% after 26 weeks at 75 µg), significance ND |
| Poewe et al., 2021 [42] | PD03A | ND | Oligomers | ND |
| Butler et al., 2022 [43] | AAV-EGFP-PFFNB2 | ↓α-syn aggregates (pS129) | Fibrils aggregates | Cortex (p < 0.001) |
| Schmidhuber et al., 2022 [44] | WISIT candidate type 1 | ↓α-syn aggregates (pS129) | Aggregates | (p < 0.05) |
| Roshanbin et al., 2022 [45] | RmAbSynO2-scFv8D3 | α-syn (total) ↓α-syn oligomers | Oligomers aggregates | No change Cortex (p < 0.05) Midbrain (p < 0.005) |
| Study | Agent | Changes in SNCA, α-Syn, TH, or Dopamine in Intervention Groups (vs. PD Model) | Location and Level of Significance for α-Syn, TH, or Dopamine |
|---|---|---|---|
| Helmschrodt et al., 2017 [46] | PEI/siRNA | ↓mRNA and α-syn protein | Striatum (medial) (p = 0.018) |
| Alarcón-Arís et al., 2018 [47] | IND-siRNA or IND-1233-ASO | ↓mRNA and α-syn protein ↑DA | SNc (p < 0.05) with IND-499-siRNA SNc (p < 0.05) with IND-1233-ASO CPu and medial PFC in response to veratridine (p < 0.05) with IND-1233-ASO |
| Izco et al., 2019 [48] | Exosomal RVG-anti-GFP shRNA-MCs | ↓α-syn protein ↑TH-labelled cells | Midbrain, 90 day treatment (p = 0.033) (p = 0.028) |
| Spencer et al., 2019 [49] | ApoB11-siRNA | ↓α-syn protein ↑TH-labelled cells | (p < 0.05) Striatum (p < 0.05) |
| Cole et al., 2021 [50] | ASO1 | ↓mRNA and α-syn aggregates ↑TH-labelled cells | SN (p < 0.001) for 100 and 300 µg dosing, (p < 0.0001) for 1000 µg dosing (p < 0.05) |
| Jin et al., 2021 [51] | Tat-βsyn-degron | ↓α-syn ↑TH-labelled cells | SN (p < 0.01) SN (p < 0.05) |
| Cao et al., 2022 [52] | HDO | ↓α-syn ↑TH-labelled cells | SN (p < 0.05) SN (p < 0.01) |
| Study | Agent | Changes in α-Syn, Dopamine, and TH in Intervention Groups (vs. PD Model) | Type of α-Syn Species Targeted | Level of Significance |
|---|---|---|---|---|
| Wagner et al., (2013) [53] | Anle138b | ↓α-syn | Oligomeric | (p < 0.001) |
| Davies et al., 2014 [54] | Nedd4 | ↓α-syn ↑TH-labelled cells | Oligomeric | (p = 0.022) (p < 0.05) |
| Savolainen et al., 2014 [55] | KYP-2047 | ↓α-syn ↑DA TH | Oligomeric | 28-d treatment: (p = 0.0028) 28-d treatment: (p = 0.01) NS |
| Wrasidlo et al., 2016 [56] | NPT100-18A | ↓α-syn | Oligomeric | (p < 0.05) |
| Kim et al., 2018 [57] | GQDs | ↓α-syn ↑TH-labelled cells | Fibrillar | (p < 0.001) (p = 0.0156) |
| Bengoa-Vergniory et al., 2020 [58] | CLR01 | ↓α-syn ↑TH-labelled cells | Oligomeric | (p = 0.0286) (p = 0.0177) |
| Xu et al., 2022 [59] | Harmol | ↓α-syn | Total | SN and PFC (p < 0.05) |
| Arotcarena et al., 2022 [60] | aNPs | α-syn (pSer129) ↑TH-labelled cells | Aggregates | NS (total/proteinase K-resistant), pSer129 (p < 0.05) (p < 0.05) |
| Kim et al., 2022 [61] | PCiv | ↓α-syn (pSer129) ↑TH-labelled cells | Aggregates | SN (p < 0.01) SN (p < 0.001) |
| Liu et al., 2022 [62] | RVG29-RBCm/Cur-NCs | ↓α-syn ↑DA ↑TH | Total | Midbrain and striatum (p < 0.01) (p < 0.001) Midbrain and striatum (p < 0.01) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodger, A.T.; ALNasser, M.; Carter, W.G. Are Therapies That Target α-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review. Int. J. Mol. Sci. 2023, 24, 11022. https://doi.org/10.3390/ijms241311022
Rodger AT, ALNasser M, Carter WG. Are Therapies That Target α-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review. International Journal of Molecular Sciences. 2023; 24(13):11022. https://doi.org/10.3390/ijms241311022
Chicago/Turabian StyleRodger, Abbie T., Maryam ALNasser, and Wayne G. Carter. 2023. "Are Therapies That Target α-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review" International Journal of Molecular Sciences 24, no. 13: 11022. https://doi.org/10.3390/ijms241311022