
Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors
 , , , , , , and
, , , , , , and 
Abstract
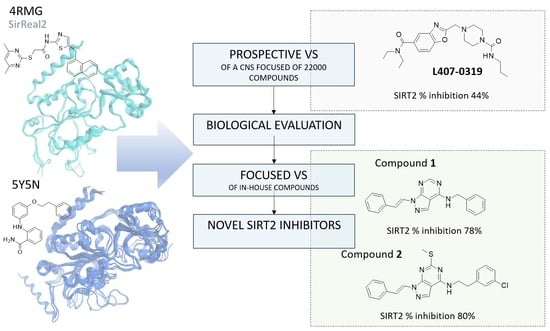
:
1. Introduction
2. Results
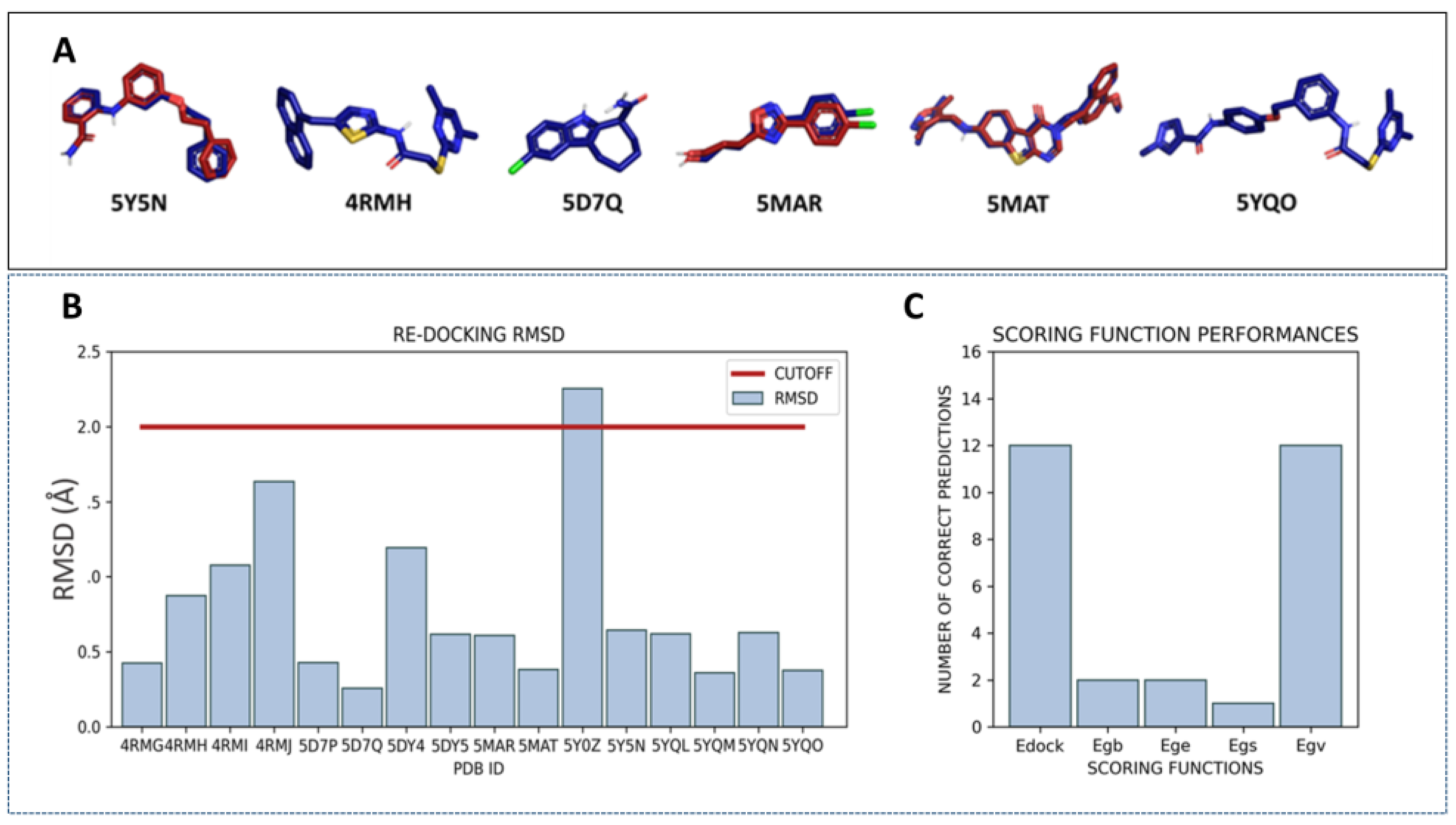
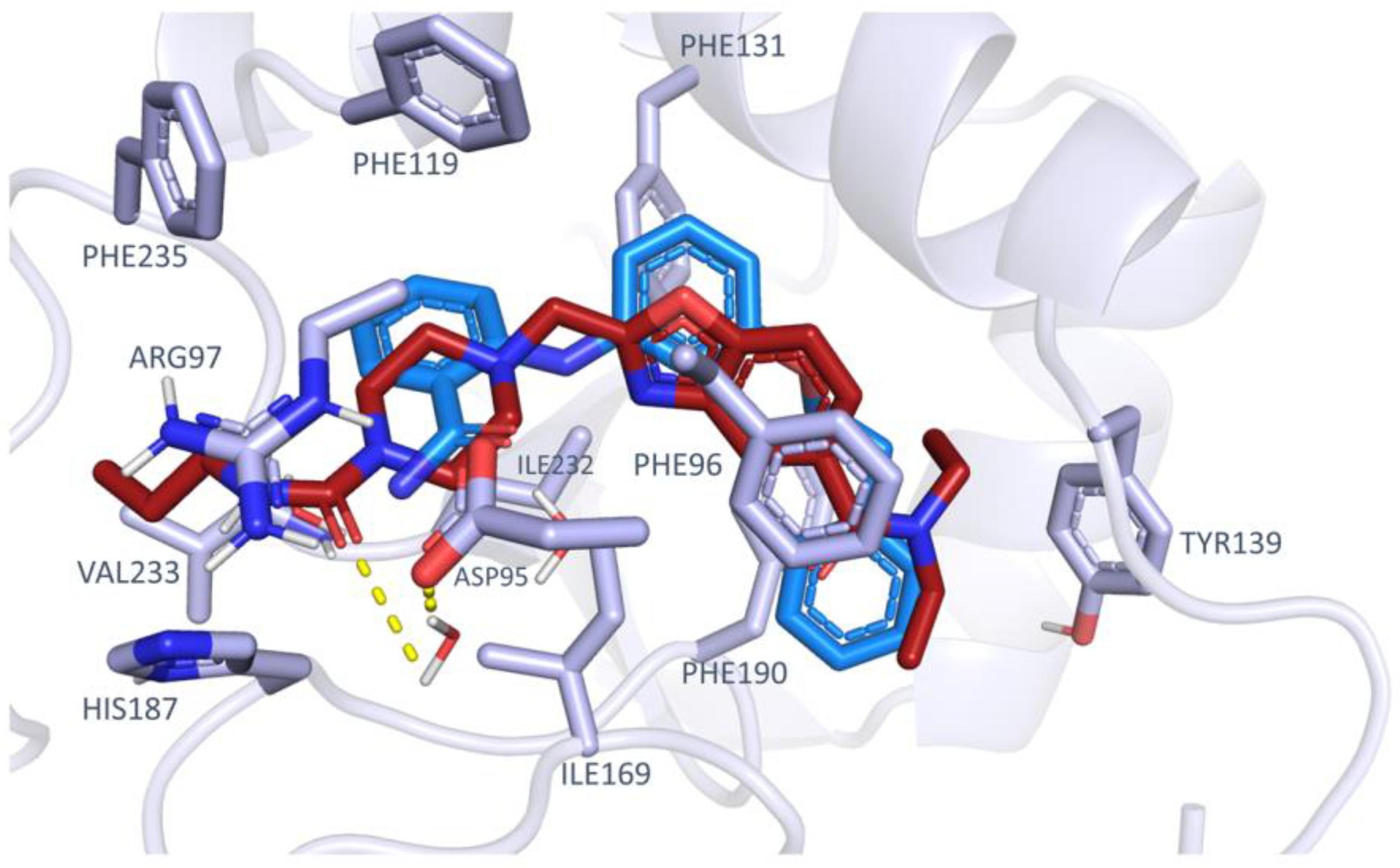
2.1. Virtual Screening Protocol Assessment
2.2. Prospective Virtual Screening of SIRT2 Inhibitors
2.3. Focused VS of Pyrazolopyrimidines as SIRT2 Inhibitors
2.4. Biochemical Assays
2.5. In Silico Prediction of ADMET Properties
3. Discussion
4. Materials and Methods
4.1. Computational Studies
4.1.1. Manual Selection of the Protein Conformations and Redocking Calculations
- Presence of substrates or peptidic inhibitors invading the binding pocket. The inclusion of these structures would prevent the positioning of the ligand in the active site, whereas their exclusion would generate a construct leaving an “empty” space which is peptide-induced, and therefore not suitable for the identification of small molecule-like inhibitors
- Presence of (peptidic) substrates with artificial groups (e.g., Trifluoro-) for the same reason described, featuring in addition artificial groups.
- Structures containing Carba-NAD. Despite the fact that it may seem a small modification, the O/C substitution is near to the binding pocket, and may influence the ligand placement.
4.1.2. Benchmarking Database Creation
4.1.3. VS Protocol Study
4.1.4. VS via Single Conformation Study
4.1.5. VS via Multi-Conformation Study
4.1.6. Induced Fit Docking
4.2. Enzymatic Assays
4.2.1. H3K9Ac and H3K9Palm Peptide Synthesis
4.2.2. Evaluation of SIRT2, SIRT1 and SIRT3 Deacetylase Activity
4.2.3. Evaluation of SIRT6 Deacetylase and Depalmitoylase Activity
4.3. In Silico Prediction of ADMET Properties
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Roshdy, E.; Mohamed, D.E.M.; Ali, T.F.S.; Gabr, G.A.; Jaragh-Alhadad, L.A.; Mekhemer, G.A.H.; Shawky, A.M.; Sidhom, P.A.; et al. In Silico Drug Discovery of SIRT2 Inhibitors from Natural Source as Anticancer Agents. Sci. Rep. 2023, 13, 2146. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-Based Development of an Affinity Probe for Sirtuin 2. Angew. Chem. Int. Ed. Engl. 2016, 55, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Osborne, B.; Bentley, N.L.; Montgomery, M.K.; Turner, N. The Role of Mitochondrial Sirtuins in Health and Disease. Free Radic. Biol. Med. 2016, 100, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kosciuk, T.; Wang, M.; Hong, J.Y.; Lin, H. Updates on the Epigenetic Roles of Sirtuins. Curr. Opin. Chem. Biol. 2019, 51, 18–29. [Google Scholar] [CrossRef]
- Salo, H.S.; Laitinen, T.; Poso, A.; Jarho, E.; Lahtela-Kakkonen, M. Identification of Novel SIRT3 Inhibitor Scaffolds by Virtual Screening. Bioorg. Med. Chem. Lett. 2013, 23, 2990–2995. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Mori, M.; Cazzaniga, G.; Meneghetti, F.; Villa, S.; Gelain, A. Insights on the Modulation of SIRT5 Activity: A Challenging Balance. Molecules 2022, 27, 4449. [Google Scholar] [CrossRef]
- Roshdy, E.; Mustafa, M.; Shaltout, A.E.R.; Radwan, M.O.; Ibrahim, M.A.A.; Soliman, M.E.; Fujita, M.; Otsuka, M.; Ali, T.F.S. Selective SIRT2 Inhibitors as Promising Anticancer Therapeutics: An Update from 2016 to 2020. Eur. J. Med. Chem. 2021, 224, 113709. [Google Scholar] [CrossRef]
- Dukanya; Shanmugam, M.K.; Rangappa, S.; Metri, P.K.; Mohan, S.; Basappa; Rangappa, K.S. Exploring the Newer Oxadiazoles as Real Inhibitors of Human SIRT2 in Hepatocellular Cancer Cells. Bioorg. Med. Chem. Lett. 2020, 30, 127330. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. Ten Years of NAD-Dependent SIR2 Family Deacetylases: Implications for Metabolic Diseases. Trends Pharmacol. Sci. 2010, 31, 212–220. [Google Scholar] [CrossRef]
- Kadam, R.U.; Tavares, J.; M, K.V.; Cordeiro, A.; Ouaissi, A.; Roy, N. Structure Function Analysis of Leishmania Sirtuin: An Ensemble of In Silico and Biochemical Studies. Chem. Biol. Drug Des. 2008, 71, 501–506. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Nebbioso, A.; Paolini, C.; Huidobro, C.; Lara, E.; Mellini, P.; Lenoci, A.; Pezzi, R.; Botta, G.; et al. Discovery of Salermide-Related Sirtuin Inhibitors: Binding Mode Studies and Antiproliferative Effects in Cancer Cells Including Cancer Stem Cells. J. Med. Chem. 2012, 55, 10937–10947. [Google Scholar] [CrossRef]
- Zhang, Y.; Au, Q.; Zhang, M.; Barber, J.R.; Ng, S.C.; Zhang, B. Identification of a Small Molecule SIRT2 Inhibitor with Selective Tumor Cytotoxicity. Biochem. Biophys. Res. Commun. 2009, 386, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Erburu, M.; Muñoz-Cobo, I.; Diaz-Perdigon, T.; Mellini, P.; Suzuki, T.; Puerta, E.; Tordera, R.M. SIRT2 Inhibition Modulate Glutamate and Serotonin Systems in the Prefrontal Cortex and Induces Antidepressant-like Action. Neuropharmacology 2017, 117, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cobo, I.; Belloch, F.B.; Díaz-Perdigón, T.; Puerta, E.; Tordera, R.M. SIRT2 Inhibition Reverses Anhedonia in the VGLUT1+/− Depression Model. Behav. Brain Res. 2017, 335, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.Q.; Zhang, P.; Tian, B.; Chen, X.Q. Downregulation of NAD-Dependent Deacetylase SIRT2 Protects Mouse Brain Against Ischemic Stroke. Mol. Neurobiol. 2017, 54, 7251–7261. [Google Scholar] [CrossRef]
- Bai, N.; Li, N.; Cheng, R.; Guan, Y.; Zhao, X.; Song, Z.; Xu, H.; Yi, F.; Jiang, B.; Li, X.; et al. Inhibition of SIRT2 Promotes APP Acetylation and Ameliorates Cognitive Impairment in APP/PS1 Transgenic Mice. Cell Rep. 2022, 40, 111062. [Google Scholar] [CrossRef]
- Abbotto, E.; Scarano, N.; Piacente, F.; Millo, E.; Cichero, E.; Bruzzone, S. Virtual Screening in the Identification of Sirtuins’ Activity Modulators. Molecules 2022, 27, 5641. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Celic, I.; Muhammad, S.; Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Structure of a Sir2 Enzyme Bound to an Acetylated P53 Peptide. Mol. Cell 2002, 10, 523–535. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the Histone Deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef]
- Li, J.; Flick, F.; Verheugd, P.; Carloni, P.; Lüscher, B.; Rossetti, G. Insight into the Mechanism of Intramolecular Inhibition of the Catalytic Activity of Sirtuin 2 (SIRT2). PLoS ONE 2015, 10, e0139095. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 Inhibition by Ligand-Induced Rearrangement of the Active Site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dubey, A.; Kamboj, N.K.; Sahoo, A.K.; Kang, S.G.; Yadava, U. Drug Repurposing for Ligand-Induced Rearrangement of Sirt2 Active Site-Based Inhibitors via Molecular Modeling and Quantum Mechanics Calculations. Sci. Rep. 2021, 11, 10169. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: https://www.rcsb.org/ (accessed on 10 January 2022).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal Structures of Human SIRT3 Displaying Substrate-Induced Conformational Changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Szczepankiewicz, B.G.; Dai, H.; Koppetsch, K.J.; Qian, D.; Jiang, F.; Mao, C.; Perni, R.B. Synthesis of Carba-NAD and the Structures of Its Ternary Complexes with SIRT3 and SIRT5. J. Org. Chem. 2012, 77, 7319–7329. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Nguyen, G.T.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A Molecular Mechanism for Direct Sirtuin Activation by Resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef]
- Huhtiniemi, T.; Salo, H.S.; Suuronen, T.; Poso, A.; Salminen, A.; Leppänen, J.; Jarho, E.; Lahtela-Kakkonen, M. Structure-Based Design of Pseudopeptidic Inhibitors for SIRT1 and SIRT2. J. Med. Chem. 2011, 54, 6456–6468. [Google Scholar] [CrossRef]
- Available online: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/cns-bbb-library/ (accessed on 1 July 2022).
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal Structure Analysis of Human Sirt2 and Its ADP-Ribose Complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef]
- Rumpf, T.; Gerhardt, S.; Einsle, O.; Jung, M. Seeding for Sirtuins: Microseed Matrix Seeding to Obtain Crystals of Human Sirt3 and Sirt2 Suitable for Soaking. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1498–1510. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Mellini, P.; Itoh, Y.; Tsumoto, H.; Li, Y.; Suzuki, M.; Tokuda, N.; Kakizawa, T.; Miura, Y.; Takeuchi, J.; Lahtela-Kakkonen, M.; et al. Potent Mechanism-Based Sirtuin-2-Selective Inhibition by an in Situ-Generated Occupant of the Substrate-Binding Site, “Selectivity Pocket” and NAD+ Binding Site. Chem. Sci. 2017, 8, 6400–6408. [Google Scholar] [CrossRef]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure–Activity Relationship, X-ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wang, H.-L.; Zhong, L.; Yuan, C.; Liu, S.-Y.; Yu, Z.-J.; Liu, S.; Yan, Y.-H.; Wu, C.; Wang, Y.; et al. X-ray Crystal Structure Guided Discovery of New Selective, Substrate-Mimicking Sirtuin 2 Inhibitors That Exhibit Activities against Non-Small Cell Lung Cancer Cells. Eur. J. Med. Chem. 2018, 155, 806–823. [Google Scholar] [CrossRef]
- Kudo, N.; Ito, A.; Arata, M.; Nakata, A.; Yoshida, M. Identification of a Novel Small Molecule That Inhibits Deacetylase but Not Defatty-Acylase Reaction Catalysed by SIRT2. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170070. [Google Scholar] [CrossRef]
- You, W.; Zheng, W.; Weiss, S.; Chua, K.F.; Steegborn, C. Structural Basis for the Activation and Inhibition of Sirtuin 6 by Quercetin and Its Derivatives. Sci. Rep. 2019, 9, 19176. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Gotoh, O. Optimal Alignment between Groups of Sequences and Its Application to Multiple Sequence Alignment. Bioinformatics 1993, 9, 361–370. [Google Scholar] [CrossRef]
- Pei, F.; Jin, H.; Zhou, X.; Xia, J.; Sun, L.; Liu, Z.; Zhang, L. Enrichment Assessment of Multiple Virtual Screening Strategies for Toll-Like Receptor 8 Agonists Based on a Maximal Unbiased Benchmarking Data Set. Chem. Biol. Drug. Des. 2015, 86, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Matsumoto, Y.; Ishikawa, M.; Sugita, K.; Hashimoto, Y.; Wakai, N.; Kitao, A.; Morishita, E.; Toyoshima, C.; Hayashi, T.; et al. Design, Synthesis and Structure–Activity Relationship Studies of Novel Sirtuin 2 (SIRT2) Inhibitors with a Benzamide Skeleton. Bioorg. Med. Chem. 2015, 23, 328–339. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Chen, W.; Song, C.; Zhang, X.; Yang, F.; Wang, C.; Zhang, Y.; Qian, S.; Wang, Z.; et al. Discovery of (5-Phenylfuran-2-Yl)Methanamine Derivatives as New Human Sirtuin 2 Inhibitors. Molecules 2019, 24, 2724. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of Potent and Selective Sirtuin 2 (SIRT2) Inhibitors Using a Fragment-Based Approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef] [PubMed]
- Khanfar, M.A.; Quinti, L.; Wang, H.; Choi, S.H.; Kazantsev, A.G.; Silverman, R.B. Development and Characterization of 3-(Benzylsulfonamido)Benzamides as Potent and Selective SIRT2 Inhibitors. Eur. J. Med. Chem. 2014, 76, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://molsoft.com/~jack/gui/tut-multiple-receptor.html (accessed on 10 January 2022).
- Chemical Computing Group ULC. Molecular Operating Environment (MOE2019.01). 2021. Available online: http://www.chemcomp.com/ (accessed on 16 January 2023).
- Carraro, F.; Naldini, A.; Pucci, A.; Locatelli, G.A.; Maga, G.; Schenone, S.; Bruno, O.; Ranise, A.; Bondavalli, F.; Brullo, C.; et al. Pyrazolo[3,4-d]Pyrimidines as Potent Antiproliferative and Proapoptotic Agents toward A431 and 8701-BC Cells in Culture via Inhibition of c-Src Phosphorylation. J. Med. Chem. 2006, 49, 1549–1561. [Google Scholar] [CrossRef]
- Tintori, C.; La Sala, G.; Vignaroli, G.; Botta, L.; Fallacara, A.L.; Falchi, F.; Radi, M.; Zamperini, C.; Dreassi, E.; Dello Iacono, L.; et al. Studies on the ATP Binding Site of Fyn Kinase for the Identification of New Inhibitors and Their Evaluation as Potential Agents against Tauopathies and Tumors. J. Med. Chem. 2015, 58, 4590–4609. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue Alpha-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel Biguanide-Based Derivatives Scouted as TAAR1 Agonists: Synthesis, Biological Evaluation, ADME Prediction and Molecular Docking Studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef]
- Parodi, A.; Righetti, G.; Pesce, E.; Salis, A.; Tasso, B.; Urbinati, C.; Tomati, V.; Damonte, G.; Rusnati, M.; Pedemonte, N.; et al. Discovery of Novel VX-809 Hybrid Derivatives as F508del-CFTR Correctors by Molecular Modeling, Chemical Synthesis and Biological Assays. Eur. J. Med. Chem. 2020, 208, 112833. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug. Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Schmidt, F.; Matter, H.; Hessler, G.; Czich, A. Predictive in Silico Off-Target Profiling in Drug Discovery. Future Sci. 2014, 6, 295–317. [Google Scholar] [CrossRef]
- Cichero, E.; Calautti, A.; Francesconi, V.; Tonelli, M.; Schenone, S.; Fossa, P. Probing in Silico the Benzimidazole Privileged Scaffold for the Development of Drug-like Anti-RSV Agents. Pharmaceuticals 2021, 14, 1307. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Bhojwani, H.R.; Joshi, U.J. Selecting Protein Structure/S for Docking-Based Virtual Screening: A Case Study on Type Ii Inhibitors of Vegfr-2 Kinase | International Journal of Pharmaceutical Sciences and Research. Int. J. Pharm. Sci. Res. 2019, 10, 2998–3011. [Google Scholar] [CrossRef]
- Cleves, A.E.; Jain, A.N. Structure- And Ligand-Based Virtual Screening on DUD-E+: Performance Dependence on Approximations to the Binding Pocket. J. Chem. Inf. Model. 2020, 60, 4296–4310. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Kumar, A.; Jangid, K.; Kumar, V.; Jaitak, V. Identification of terpenoids as dihydropteroate synthase and dihydrofolate reductase inhibitors through structure-based virtual screening and molecular dynamic simulation. J. Biomol. Struct. Dyn. 2023, 1–19. [Google Scholar] [CrossRef]
- Djokovic, N.; Ruzic, D.; Rahnasto-Rilla, M.; Srdic-Rajic, T.; Lahtela-Kakkonen, M.; Nikolic, K. Expanding the Accessible Chemical Space of SIRT2 Inhibitors through Exploration of Binding Pocket Dynamics. J. Chem. Inf. Model. 2022, 62, 2571–2585. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Yang, W.; Chen, W.; Su, H.; Li, R.; Song, C.; Wang, Z.; Yang, L. Recent Advances in the Development of Histone Deacylase SIRT2 Inhibitors. RSC Adv. 2020, 10, 37382–37390. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE2019.01): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from the Distorted Native Conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Halim, S.A.; Ul-Haq, Z.; Khan, A.; Al-Rawahi, A.; Al-Harrasi, A. In silico data mining of large-scale databases for the virtual screening of human interleukin-2 inhibitors. Acta Pharm. 2021, 71, 33–56. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Rapetti, F.; Lusardi, M.; Scarano, N.; Alfei, S.; Altieri, P.; Garibaldi, S.; Ameri, P.; Grazia Signorello, M.; Brullo, C. Scouting Different Phosphodiesterase 4 Inhibitor Chemotypes in Silico To Guide the Design of Anti-inflammatory/Antioxidant Agents. ChemMedChem 2023, 18, e202300046. [Google Scholar] [CrossRef] [PubMed]
- Sociali, G.; Liessi, N.; Grozio, A.; Caffa, I.; Parenti, M.D.; Ravera, S.; Tasso, B.; Benzi, A.; Nencioni, A.; Del Rio, A.; et al. Differential Modulation of SIRT6 Deacetylase and Deacylase Activities by Lysine-Based Small Molecules. Mol. Divers. 2020, 24, 655–671. [Google Scholar] [CrossRef]
- Sociali, G.; Galeno, L.; Parenti, M.D.; Grozio, A.; Bauer, I.; Passalacqua, M.; Boero, S.; Donadini, A.; Millo, E.; Bellotti, M.; et al. Quinazolinedione SIRT6 Inhibitors Sensitize Cancer Cells to Chemotherapeutics. Eur. J. Med. Chem. 2015, 102, 530–539. [Google Scholar] [CrossRef]
- ACD/Percepta Platform; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2015.
- Acdlabs. Available online: www.acdlabs.com (accessed on 1 July 2022).
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the Interaction Landscape of Bioactive Molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
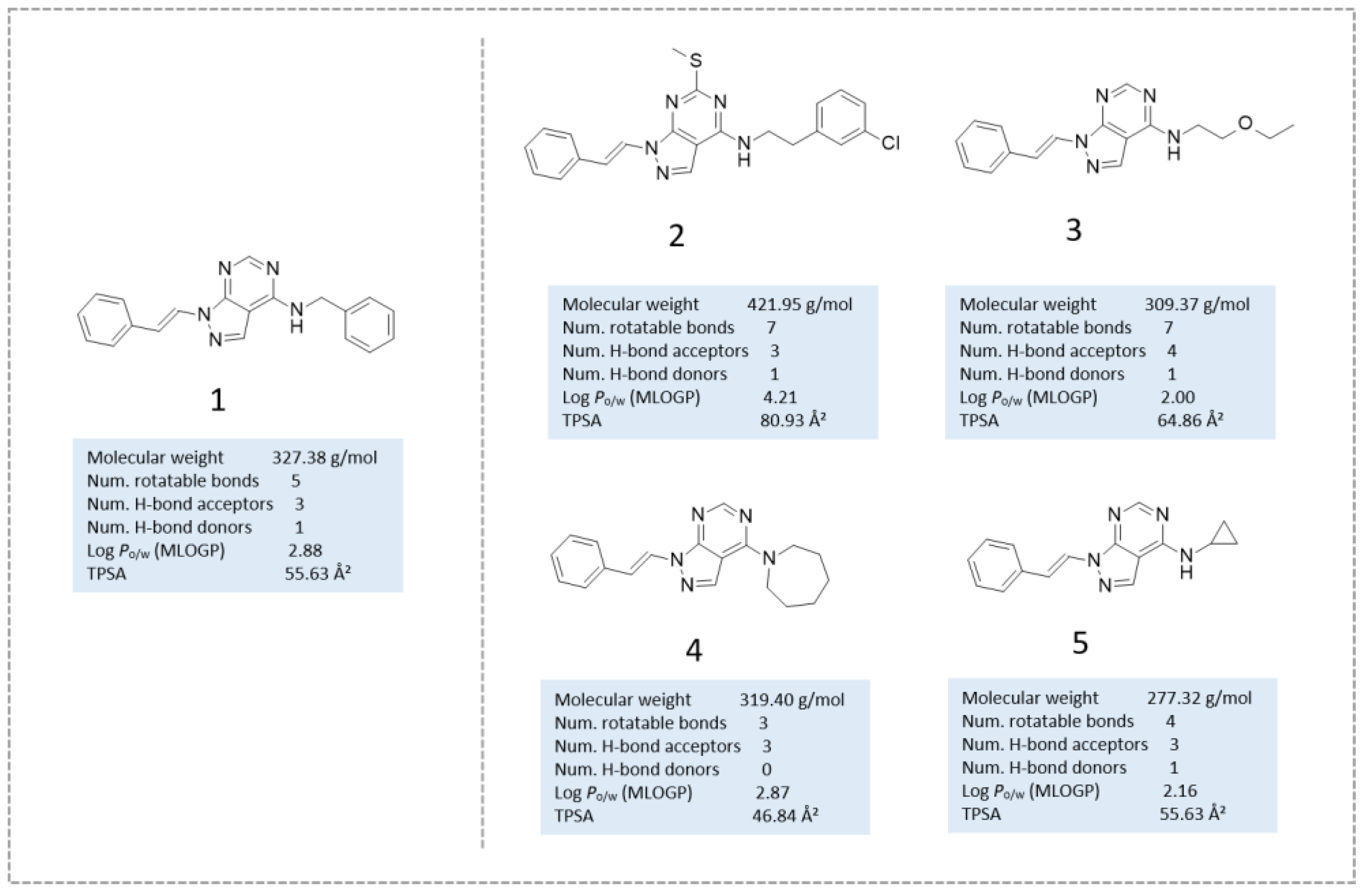{kind=link}
{kind=link}
| Entry | PDB ID | Inhibitor | Cofactor | Substrate | Resolution (Å) | Ref. | Sequence Length | Year |
|---|---|---|---|---|---|---|---|---|
| 1 | 1J8F | None | None | None | 1.70 | [20] | 323 | 2001 |
| 2 | 3ZGV | None | ADPR | None | 2.27 | [31] | 325 | 2013 |
| 3 | 3ZGO | none | none | None | 1.63 | [31] | 325 | 2013 |
| 4 | 4RMG | SirReal2 | NAD | None | 1.88 | [22] | 304 | 2015 |
| 5 | 4RMH | SirReal2 | None | Ac-Lys-H3 peptide | 1.42 | [22] | 304 | 2015 |
| 6 | 4RMI | SirReal1 | None | Ac-Lys-OTC peptide | 1.45 | [22] | 304 | 2015 |
| 7 | 4RMJ | Nicotinamide | ADPR | none | 1.87 | [22] | 304 | 2015 |
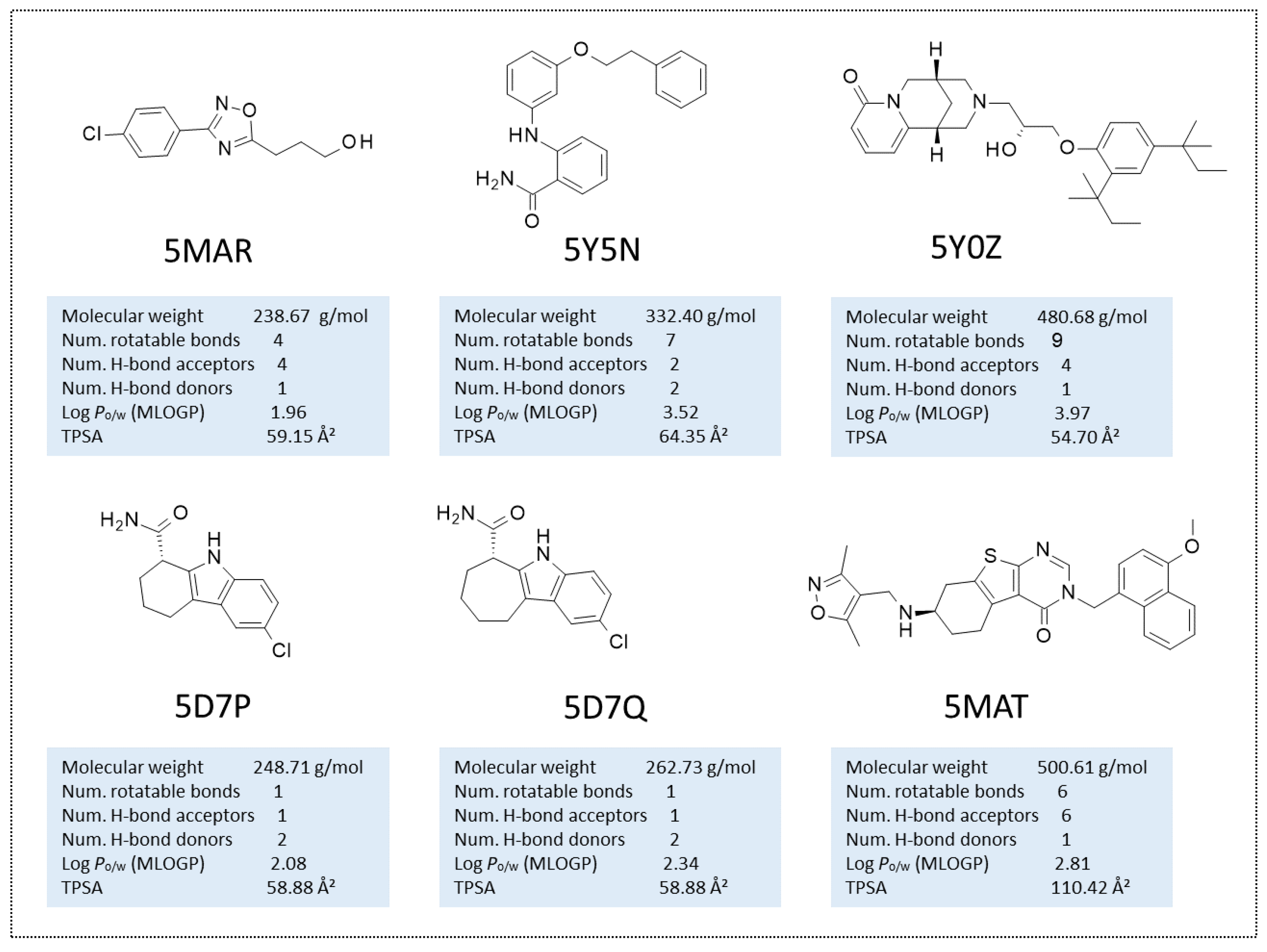
| 8 | 5D7P | EX-243 | ADPR | None | 1.76 | [32] | 304 | 2015 |
| 9 | 5D7O | none | ADPR | None | 1.63 | [32] | 310 | 2015 |
| 10 | 5D7Q | CHIC35 | ADPR | None | 2.01 | [32] | 304 | 2015 |
| 11 | 5DY4 | SirReal analog | NAD | None | 1.77 | [33] | 304 | 2016 |
| 12 | 5DY5 | SirReal probe | None | None | 1.95 | [2] | 304 | 2016 |
| 13 | 5Y5N | Small molecule-inhibitor | None | None | 2.30 | [34] | 336 | 2017 |
| 14 | 5MAR | oxadiazole | ADPR | None | 1.89 | [35] | 303 | 2017 |
| 15 | 5MAT | Thienopyrimidinone | None | None | 2.07 | [35] | 303 | 2017 |
| 16 | 5YQL | A2I | None | None | 1.60 | [36] | 306 | 2018 |
| 17 | 5YQM | A29 | None | None | 1.74 | [36] | 306 | 2018 |
| 18 | 5YQN | L55 | None | None | 1.60 | [36] | 306 | 2018 |
| 19 | 5YQO | L5C | None | None | 1.48 | [36] | 306 | 2018 |
| 20 | 5Y0Z | NPD11033 | None | None | 2.00 | [37] | 293 | 2018 |
| 21 | 6QCN | Quercetin (out of the protein) | ADPR | None | 2.23 | [38] | 304 | 2019 |
| Molecule | Activity (IC50, %inhib) | SMILE Notation | References |
|---|---|---|---|
 | 0.90 μM | O=C(Nc1cccnn1)c2cccc(c2)c3ccc4ccccc4c3 | [42] |
 | 1.74 μM | NC(=O)c1ccccc1Nc2cccc(OCCc3ccccc3)c2 | [34] |
 | 99% | OC(=O)c1ccc(cc1)c2oc(CNC(=O)Nc3cccnc3)cc2 | [43] |
 | 1.5 μM | BrCCCc1onc(n1)c2ccc(Br)cc2 | [35] |
 | 0.0483 μM | NC(=O)c1cncc(Oc2cccc3c(NC(=O)c4ccccc4)cccc23)c1 | [44] |
 SirReal2 SirReal2 | 0.4 μM | Cc1cc(C)nc(SCC(=O)Nc2ncc(Cc3cccc4ccccc34)s2)n1 | [22] |
 | 4.9 μM | CN(c1ccc(Cl)cc1)S(=O)(=O)c2cc(ccn2)C(=O)Nc3ccc(cc3)C#N | [45] |
 3-AGK2 3-AGK2 | 1.56 μM | Clc1ccc(Cl)c(c1)c2oc(\C=C(/C#N)\C(=O)Nc3cccc4ncccc34)cc2 | [44] |
 | 3.5 μM | [O-][N+](=O)c1ccc(Sc2nnc(COc3ccccc3)n2c4ccccc4)c5ncccc15 | [46] |
 | 0.815 μM | Cc1cc(C)nc(SCC(=O)Nc2cccc(COc3ccc(NC(=O)c4cnn(C)c4)cc3)c2)n1 | [36] |
| Molecule | SIRT2 Activity, %Inhibition (at 150 μM) |
|---|---|
| L407-0319 | 44.3 ± 5.3 |
| L929-0391 | 24.4 ± 6.2 |
| S787-1020 | NI |
| T158-0512 | NI |
| G779-0661 | 25.2 ± 5.9 |
| 1 | 81.2 ± 7.3 |
| 2 | 79.9 ± 5.7 |
| 3 | 51.1 ± 15.2 |
| 4 | 35.7 ± 2.5 |
| 5 | 31.4± |
| AGK2 | 97 ± 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarano, N.; Abbotto, E.; Musumeci, F.; Salis, A.; Brullo, C.; Fossa, P.; Schenone, S.; Bruzzone, S.; Cichero, E. Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors. Int. J. Mol. Sci. 2023, 24, 9363. https://doi.org/10.3390/ijms24119363
Scarano N, Abbotto E, Musumeci F, Salis A, Brullo C, Fossa P, Schenone S, Bruzzone S, Cichero E. Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors. International Journal of Molecular Sciences. 2023; 24(11):9363. https://doi.org/10.3390/ijms24119363
Chicago/Turabian StyleScarano, Naomi, Elena Abbotto, Francesca Musumeci, Annalisa Salis, Chiara Brullo, Paola Fossa, Silvia Schenone, Santina Bruzzone, and Elena Cichero. 2023. "Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors" International Journal of Molecular Sciences 24, no. 11: 9363. https://doi.org/10.3390/ijms24119363