
Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway


Department of Clinical Research and Innovation (DRCI), Foch Hospital, 92150 Suresnes, France
Int. J. Mol. Sci. 2022, 23(5), 2810; https://doi.org/10.3390/ijms23052810
Submission received: 26 January 2022
/
Revised: 24 February 2022
/
Accepted: 27 February 2022
/
Published: 4 March 2022
(This article belongs to the Special Issue New Advance in Neuroinflammation)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Schizophrenia is a very complex syndrome involving widespread brain multi-dysconnectivity. Schizophrenia is marked by cognitive, behavioral, and emotional dysregulations. Recent studies suggest that inflammation in the central nervous system (CNS) and immune dysfunction could have a role in the pathogenesis of schizophrenia. This hypothesis is supported by immunogenetic evidence, and a higher incidence rate of autoimmune diseases in patients with schizophrenia. The dysregulation of the WNT/β-catenin pathway is associated with the involvement of neuroinflammation in schizophrenia. Several studies have shown that there is a vicious and positive interplay operating between neuroinflammation and oxidative stress. This interplay is modulated by WNT/β-catenin, which interacts with the NF-kB pathway; inflammatory factors (including IL-6, IL-8, TNF-α); factors of oxidative stress such as glutamate; and dopamine. Neuroinflammation is associated with increased levels of PPARγ. In schizophrenia, the expression of PPAR-γ is increased, whereas the WNT/β-catenin pathway and PPARα are downregulated. This suggests that a metabolic-inflammatory imbalance occurs in this disorder. Thus, this research’s triptych could be a novel therapeutic approach to counteract both neuroinflammation and oxidative stress in schizophrenia.
Keywords:
neuroinflammation; oxidative stress; schizophrenia; WNT/β-catenin pathway; glutamate; PPARγ; PPARα1. Introduction
Schizophrenia is a neurodevelopmental and mental illness affecting nearly 1% of the world’s population, and the age of onset age is between 23 and 34 years in women and close to 30 years in men [1]. Schizophrenia is characterized by generalized brain multi-dysconnectivity and marked by abnormal brain development, deregulated neuronal migration, impaired spatial neural arrangement, and the absence of gliosis [2]. This mental disorder is composed of cognitive, behavioral, and emotional disorders, and to be diagnosed with schizophrenia, patients should have two (or more) of the following negative symptoms for a minimum of six months—disorganized speech, grossly disorganized or catatonic behavior—and at least one positive symptom [3]. Positive symptoms are composed of delusions and hallucinations, while negative symptoms are deficits in normal behavior, such as alogia, associability, blunted affect, or anhedonia [4]. The pathophysiological process of the onset of schizophrenia remains unknown to this day. Many signaling pathways have been researched in attempts to explain the neurodevelopmental pathology of this disorder, with genetic, neurodevelopmental, and neurochemical hypotheses being proposed [5]. One possible hypothesis postulates that schizophrenia may be caused by changes (i.e., neuroinflammation and oxidative stress) in the fetus. However, this hypothesis remains both uncertain and unclear.
Recent investigations have observed that the inflammation of the central nervous system (CNS) and immune dysfunction may have a role in the pathogenesis of schizophrenia. This hypothesis developed from immunogenetic evidence and the high propensity of schizophrenic patients to develop autoimmune diseases [6,7]. Neuroinflammation can cause white matter damage and dysconnectivity, therefore generating the appearance of symptoms related to schizophrenia [6].
The WNT/β-catenin pathway is mainly involved in the control of cell development and regeneration, as well as tissue homeostasis [8,9]. This pathway is also associated with several other psychiatric diseases and disorders [10,11,12,13]. The disruption of the WNT/β-catenin pathway is associated with the occurrence of chronic neuroinflammation in several diseases, in particular schizophrenia [11,14].
Understanding the complexity of the relationship between the WNT/β-catenin pathway and neuroinflammation in schizophrenia will help to identify new therapeutic targets. Thus, this review focuses on describing the main role of neuroinflammation in schizophrenia and its relationship with the WNT/β-catenin pathway, which leads to the development of oxidative stress, and highlighting possible therapeutic targets.
2. Neuroinflammation in Schizophrenia
The interrelationship between the immune and neuroinflammatory systems plays a major role in the neurobiology of schizophrenia [15]. Recent research has shown that chronic neuroinflammation affecting the CNS could be associated with schizophrenia [16]. Indeed, a significant association was observed between schizophrenia and the expression of the major histocompatibility complex (MHC), located on chromosome 6, in brain and immune cells involved in adaptive immunity (CD19 and CD20B lymphocytes) [17]. Moreover, the overstimulation of dopamine D3 receptors and interferon γ synthesis (IFNγ) in lymphocytes has been observed in unmedicated schizophrenic patients [18].
Several studies, including meta-analyses, have found immune dysfunction in schizophrenia [19,20]. In contrast, other studies have observed immunological modification in only 40% of schizophrenic patients [21,22]. It remains important to define efficient immune biomarkers to identify schizophrenic patients who could benefit from anti-inflammatory treatment [23]. In schizophrenic disorder, several studies have shown that the secretion of pro-inflammatory and anti-inflammatory cytokines is concomitantly disrupted [24]. Cytokines represent a wide range of molecules generated by cells, such as B and T immune cells, lymphocytes, macrophages, endothelial cells, and fibroblasts. In the first psychotic episode of schizophrenia, a decrease in anti-inflammatory markers, such as interleukins IL-10 and IL-4, has been observed, while pro-inflammatory factors, such as IL-6, are increased [1]. The oversecretion of cytokines has also been observed in peripheral blood mononuclear cells, with the stimulation of the expression of IL-6, IL-8, and tumor necrosis factor α (TNF-α) and a decreased expression of IL-2mRNA [21]. In the cerebrospinal fluid (CSF) of schizophrenic patients, the dysregulation in the expression of cytokines has also been observed [25], with increased levels of IL-1, IL-6, and IL-8 [17]. In addition, a relationship has been observed between the levels of IL-6, TNF-α, and IFNγ; the high risk of subclinical psychotic symptoms with a marked decrease in their expression under antipsychotic treatment has also been seen [17].
Many inflammatory endothelial cell molecules are involved in schizophrenic disorders, including creatine kinase m/B, angiotensin-converting enzyme, matrix metalloproteinase, thyroid-stimulating hormone, thyroxine-binding globulin, intercellular adhesion molecule 1, cortisol, macroglobulin α-2, and thrombopoietin. The altered expression of these molecules contributes to the activation of the acute phase response, leading to the stimulation of the coagulation/fibrinolytic system, which affects vascular permeability and nitric oxide production by endothelial cells [26]. This result suggests a possible peripheral molecular signature associated with immune function in early-onset schizophrenia patients. Moreover, according to the results of a meta-analysis focused on schizophrenia, absolute blood counts revealed a significant increase in total lymphocytes, CD3, and CD4, as well as in the CD4/CD8 ratio. CD3% was decreased in drug-native first-episode psychosis. Increased levels in CD4% and CD56% were observed in acutely relapsed subjects. These findings could be relevant in targeting a blood lymphocyte dysregulation signature for the initiation of antipsychotic medications [27].
C-Reactive Protein (CRP) is one of the main markers of inflammation. CRP modulation is a major marker of therapeutic response for anti-inflammatory treatment [19,28]. CRP is synthesized in the liver and released by macrophages and adipocytes in response to the stimulation of the expression of IL-1β, IL-6, and TNF-α [19]. Symptoms of schizophrenia and cognitive dysfunction are strongly associated with an increase in CRP [29,30]. Moreover, resistance to the treatment of schizophrenia is correlated with high levels of CRP [31].
The oxidative stress observed in schizophrenia is associated with the secretion of pro-inflammatory cytokines, leading to a chronic neuroinflammatory process and genetic damage that in turn promotes chronic inflammation, surrounding oxidative stress impacting cells, and immune impairment [1]. A vulnerability–stress–inflammation interaction model operates in schizophrenia. This model of schizophrenia includes the contribution of oxidative stress as a cause of increased genetic vulnerability in this pathogenesis. Oxidative stress can increase the secretion of pro-inflammatory cytokines, leading to a lasting pro-inflammatory state. Thus, the vulnerability–stress–inflammation relationship observed in schizophrenia may suggest that inflammatory response in the mother can induce genetic damages during pregnancy. This damage leads to deleterious effects on the neurological development of the fetus and can increase the risk of developing schizophrenia. Nevertheless, this hypothesis remains very uncertain to this day.
The association between oxidative stress and chronic inflammation increases the release of pro-inflammatory cytokines, the stimulation of astrocyte activity, dopaminergic and glutamatergic pathway dysregulation, and finally the onset of schizophrenia symptoms. The oxidative stress seen in schizophrenia interacts with inflammation [32], suggesting that the symptoms of schizophrenia are associated with specific changes in dopaminergic, serotonergic, noradrenergic, and glutamatergic neurotransmission. The dysfunction of the immune system can hinder the neurotransmission of dopamine and glutamate. The immune system can stimulate indoleamine 2,3-dioxygenase, an enzyme involved in the tryptophan/kynurenine mechanism [15]. In the brain, kynurenic acid acts as a natural antagonist of N-methyl-D-aspartate (NMDA). In the CSF of schizophrenic patients, kynurenic acid levels are increased [33]. Pro-inflammatory cytokines stimulate the concentration of kynurenic acid and lead to the production of antibodies to the NMDA receptor. This process supports the idea that the involvement of the glutamatergic pathway and NMDA receptor is major in schizophrenic patients [15,32].
3. The WNT/β-Catenin Pathway
The name WNT (Wingless-related integration site) is derived from Wingless drosophila melanogaster and its mouse homolog Int. The WNT/β-catenin pathway is involved in many regulatory pathways, such as embryogenesis, cell proliferation, migration and polarity, apoptosis, and organogenesis [34]. However, in several pathological conditions, the WNT/β-catenin pathway is disrupted, such as in inflammatory, metabolic, and neurological disorders; tissue fibrosis; and cancers [13,35,36].
The WNT proteins are members of the family of glycoproteins produced by modified lipids [37]. WNT ligands are produced by neurons and immune cells in the CNS [38]. The regulation of the WNT/β-catenin pathway leads to the involvement of embryonic development, cell fate, epithelial-mesenchymal transition (EMT), and metabolism. The formation of the complex composed by the transcriptional coactivator β-catenin and its transcriptional factor T Cell Factor/Lymphoid Enhancer Factor (TCF/LEF) is considered as a major step of the WNT/β-catenin pathway. The accumulation of β-catenin in the cytosol is modulated by the formation of the destruction complex composed of AXIN, glycogen synthase kinase-3 (GSK-3β), tumor suppressor adenomatous polyposis (APC), protein phosphatase 2A (PP2A), and casein kinase 1α CK1α. In the absence of WNT ligands, the destruction complex hyper-phosphorylates the cytosolic β-catenin and promotes its proteasomal degradation. In contrast, if they are present, WNT ligands complex with Frizzled (FZL) and low-density lipoprotein (LDL) receptor-bound protein 5/6 (LRP 5/6) to decrease the action of the β-catenin destruction complex. Thus, β-catenin transfers to the nucleus to bind to the TCF/LEF. This mechanism activates the different WNT target genes [39,40,41]. GSK-3β is a main inhibitor of the WNT/β-catenin pathway [42,43,44,45,46,47]. It is involved in the modulation of many pathophysiological pathways, such as cell membrane signaling, cell polarity, and inflammation [48,49,50].
3.1. WNT/β-Catenin Pathway and Schizophrenia
As mentioned previously, WNT/β-catenin pathway dysregulation leads to many deleterious effects in neuronal development, contributing to the pathogenesis of neurodevelopmental diseases, such as schizophrenia [51,52]. More specifically, a recent study indicated that the WNT/β-catenin pathway plays a major role in modulating dopaminergic (DA) activities [53]. The inhibition of GSK-3β activity and increased cytosolic accumulation of β-catenin promote the transformation of neuronal precursors into dopaminergic neurons, demonstrating that the WNT/β-catenin pathway can regulate neural fate in DA activity [54]. Furthermore, several authors have observed that the levels of β-catenin, APC, and the activity of GSK-3β are deregulated in the hippocampus of schizophrenic mice models [55,56,57]. Moreover, the WNT and FZD ligands are damaged in schizophrenia [58,59,60]. A genome-wide single-nucleotide polymorphism study showed that the deregulation of the membrane expression of FZD was associated with the onset of schizophrenia [61]. However, while FZD3 has been proposed as a possible marker of schizophrenic susceptibility [62,63], other studies have failed to support this hypothesis [64]. Nevertheless, these results may suggest that the disruption of the WNT/β-catenin pathway plays a major role in schizophrenia and may be a targeted pathway for its treatment.
3.2. Interplay between WNT/β-Catenin Pathway and Inflammatory Markers
The activation of the nuclear factor-kappa B (NF-κB) pathway and stimulation of the expression of cytokines and prostaglandins are responsible, in part, for neuroinflammation [65,66]. Numerous studies have shown the close relationship between the NF-κB pathway and the WNT/β-catenin pathway. This relationship operates through a negative interaction between the WNT/β-catenin and the NF-κB pathways. The WNT/β-catenin pathway decreases the activity of the NF-κB pathway [67,68,69], which controls several diverse targets, such as cytokines, chemokines, growth factors, immune receptors, transcription factors, and apoptosis repressors [70]. The NF-κB pathway is a key component of both acute and chronic inflammation. This pathway is composed of cytosolic-related transcription factors of two proteins (homodimers or heterodimers) of the Rel family of proteins, which are retained in the cytosol by an inhibitory protein called I-κB. The phosphorylation of I-κB causes the degradation of the inhibitor, leading to the release of NF-κB for translocation into the nucleus to activate its target genes. The activation of β-catenin signaling is associated with the inhibition of NF-κB pathway activity via NF-κB DNA binding arrest [70]. Other studies have shown that the inhibition of the GSK-3β activity leads to reduced activity of the NF-κB pathway [71]. GSK-3β activates the NF-κB pathway but decreases the level of β-catenin at the cytosolic level [72,73]. Thus, the activation of β-catenin, either directly or via the inhibition of GSK-3β, is associated with the reduction in NF-κB pathway activity and results in a decrease in target genes, including IL-6, IL-8, and TNF-α. This negative relationship may show a possible anti-inflammatory role of the WNT/β-catenin pathway [68,74].
To this effect, β-catenin signaling can complex with RelA and p50 to stop the NF-κB DNA binding and its transactivation activity. However, the protein–protein relationship between β-catenin and the NF-κB pathway is indirect. These two pathways do not directly bind to each other, as stimulated β-catenin decreases the expression of the NF-κB target gene named FAS [75]. Similarly, other studies focused on colorectal cancer cells have shown that stimulated β-catenin inhibits the NF-κB pathway through the activation of the phosphatidylinositide 3-kinase (PI3K) pathway [76]. This relationship is also observed in non-tumor cells, such as chondrocytes, fibroblasts, epithelial cells, osteoblasts, and hepatocytes [71].
3.3. Interplay between WNT/β-Catenin Pathway and Neuroinflammation in Schizophrenia
The disruption of the WNT/β-catenin pathway is observed in several diseases associated with chronic neuroinflammation, such as neurodegenerative diseases, but also in psychotic disorders, such as obsessive-compulsive disorder, depression, and autism spectrum disorder [8,11,77,78,79,80]. This deregulation is mainly observed in the cells of the CNS, such as macrophages/microglia, astrocytes, and oligodendrocytes. This suggests that the WNT/β-catenin pathway could be targeted to repair brain damage induced by neuroinflammation.
The inflammatory process stimulates peripheral immunoinflammatory pathways, such as IL-6, high-mobility box protein 1 (HMGB1), Dickkopf 1 (DKK1), and CCL11 (eotaxin) [81,82]. Thus, an increase in the expression of IL-6 leads to the overstimulation of both HMGB1 and DKK1 in schizophrenia [82]. Only when the WNT/β-catenin pathway is decreased can the neuroinflammatory process operate [81,83]. In parallel with the overstimulation of IL-6 and the decrease in IL-10, the disruption of the WNT/β-catenin pathway leads to the initiation of the neurotoxicity process in schizophrenic patients [82,84].
4. Interplay between Neuroinflammation and Oxidative Stress in Schizophrenia
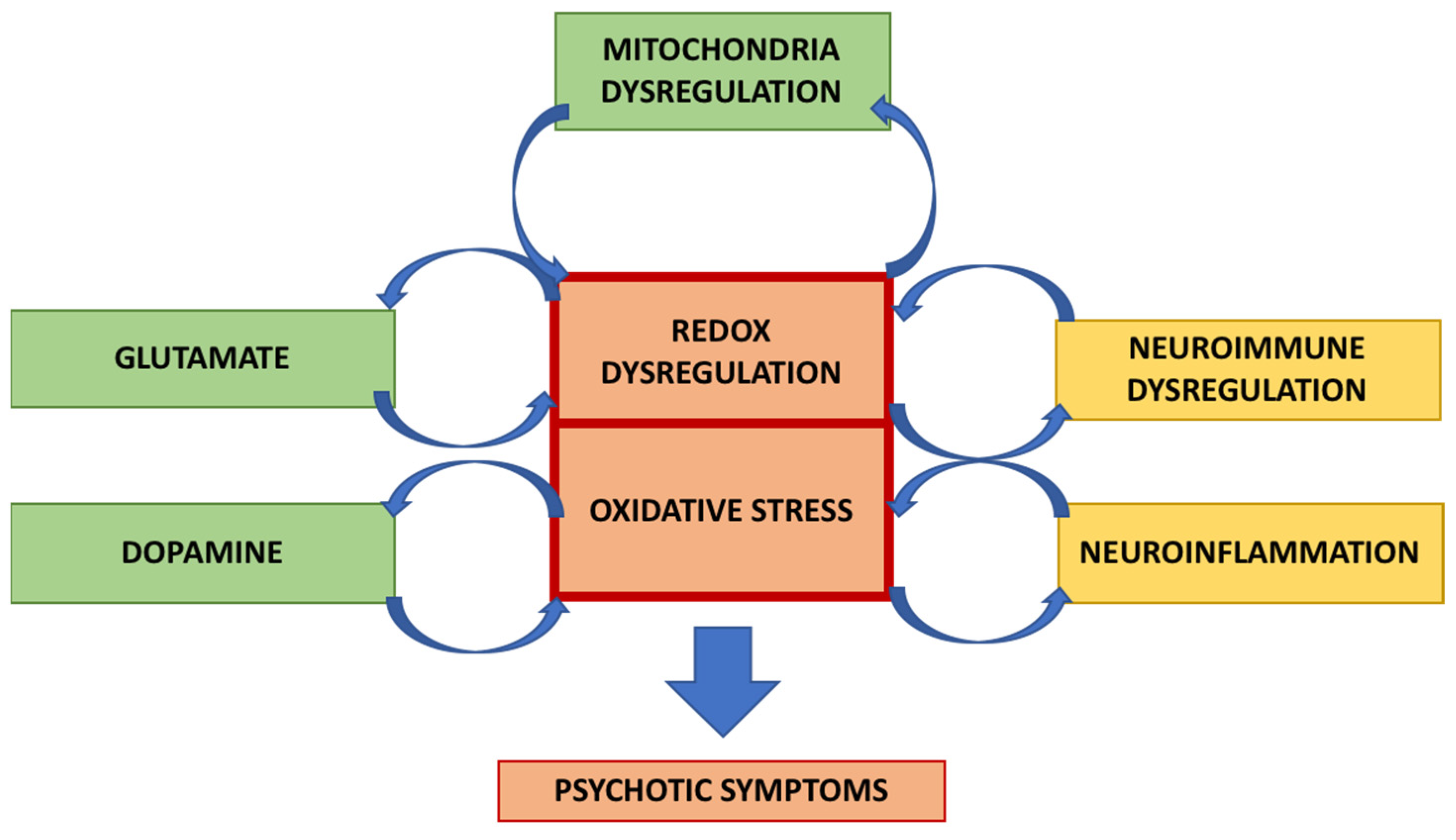
Previous studies have shown that a vicious and positive interplay operates between oxidative stress and neuroinflammation (Figure 1).
Indeed, the activation of the NF-κB pathway leads to the production of prostaglandins and the development of oxidative stress [85]. Similarly, in a direct feedback, oxidative stress can activate the NF-κB pathway. Several inflammatory proteins, such as cyclooxygenase 2 (COX-2) and matrix metalloproteinase-9 (MMP-9), are stimulated by pro-inflammatory cytokines through reactive oxygen species (ROS)-dependent signaling in astrocytes [86].
A strong relationship between oxidative stress and the immune system has been shown to be involved in schizophrenia [1]. Glutathione (GSH), an antioxidant that is essential for the myelination and maturation of white matter, is stimulated by the amino acid precursor N-acetyl cysteine (NAC). NAC has many properties: antioxidant, anti-inflammatory, and the control of NMDA synaptic receptors. NAC supplementation improves positive symptoms and neurocognition in schizophrenia patients with high peripheral oxidative stress [87]. Omega-3 polyunsaturated fatty acids also exhibit antioxidant capacity and anti-inflammatory effects [88].
Pro-inflammatory cytokines are associated with the schizophrenic process via the disruption of major neurotransmitter systems [89]. Pro-inflammatory cytokines are associated with increased concentrations of kynurenic acid, a natural antagonist of NMDA receptors. Kynurenic acid is responsible for the inhibition of the NMDA receptor. The disruption in glutamatergic transmission leads to symptoms of schizophrenia [90]. Thus, neuroinflammation and oxidative stress are mainly interdependent. Indeed, tissue damage caused by oxidative stress could be directly responsible for neuroinflammation and immune response [91]. Macrophages and microglia use ROS to destroy pathogens [92]. This observation suggests that oxidative stress may be both the cause and the result of neuroinflammation [93]. Oxidative stress activates the NF-κB pathway, which leads to an increased production of more free radicals [94]. However, the immune system is primarily a source of oxidative stress due to the activation of microglia using NADPH oxidase to produce reactive superoxide to destroy pathogens [95].
5. Oxidative Stress in Schizophrenia
Oxidative stress is a well-known process in schizophrenia [84,96]. Although this may not be the leading cause of schizophrenia, new results have suggested that oxidative stress may participate in the declining course of schizophrenia [97].
A large number of studies have evaluated peripheral biomarkers of oxidative stress, including antioxidant levels. Recent studies have observed that oxidative stress biomarkers, such as GSH, are at low levels in the plasma of schizophrenic patients [98,99]. In contrast, high levels of ROS have been found in schizophrenic patients [100]. The increase in ROS is strongly correlated with a decrease in superoxide dismutase (SOD) and glutathione peroxidase (GPx) levels [101]. The phases of the disorder, acute or stable, modulate the redox regulation [102]. Post-mortem studies have shown that there is a reduction in GSH levels in the prefrontal cortex and caudate of schizophrenic patients [103,104], showing that oxidative stress is expressed via the abnormal expression of GSH in the anterior cingulate cortex (ACC) [105]. Decreased levels of GSH in the blood are associated with the onset of symptoms of psychosis and alterations in brain volume in schizophrenia [106]. Moreover, as previously discussed, the administration of NAC in combination with antipsychotic therapies could soothe the symptoms of schizophrenia [107].
Furthermore, redox dysfunction leading to oxidative stress in schizophrenia [102,108] is characterized by the low expression of polyunsaturated fatty acids (PUFAs) in red blood cells during the acute phase of schizophrenia [108]. This process is also observed during the stable phase of schizophrenia, with a high expression of 2-amino butyrate in the low PUFA group showing persistent redox dysregulation [102]. Thus, the process of oxidative stress leads to neuronal excitability through the disruption of mitochondrial functions in schizophrenia [109]. To this end, the deficiency in antioxidant defense in schizophrenic patients could be associated with the appearance of aggravated symptoms [110].
6. The Glutamate Hypothesis of Oxidative Stress in Schizophrenia
Rapid excitatory neurotransmission is modulated in the CNS by glutamate. In neurons, glutamate is stored in synaptic vesicles, from which it is released. The release of glutamate leads to a sharp increase in its concentration in the synaptic cleft, which connects the ionotropic glutamate receptors. Glutamate is then removed from the synaptic cleft to be transported to the astrocytes by glutamate transporters (such as GLT-1 or excitatory amino acid transporters 1 and 2: EAATs 1 and 2) to prevent the upregulation of glutamate receptors [111]. Astrocytes remove more than 90% of excess glutamate by EAATs and play a major role in the glutamate/glutamine cycle. After glutamate absorption, glutamine synthetase (GS) catalyzes the ATP-dependent reaction of glutamate and ammonia into glutamine. Glutamine is released and, in turn, is absorbed by neurons to be converted into glutamate by glutaminase.
Schizophrenic patients may present with low levels of glutamate in the CSF [112]. This hypothesis of the role of glutamate in schizophrenia could indicate that the negative symptoms observed are partly caused by a dysfunctional glutamatergic pathway, and thus are directly modulated by NMDA receptors on GABAergic interneurons [113].
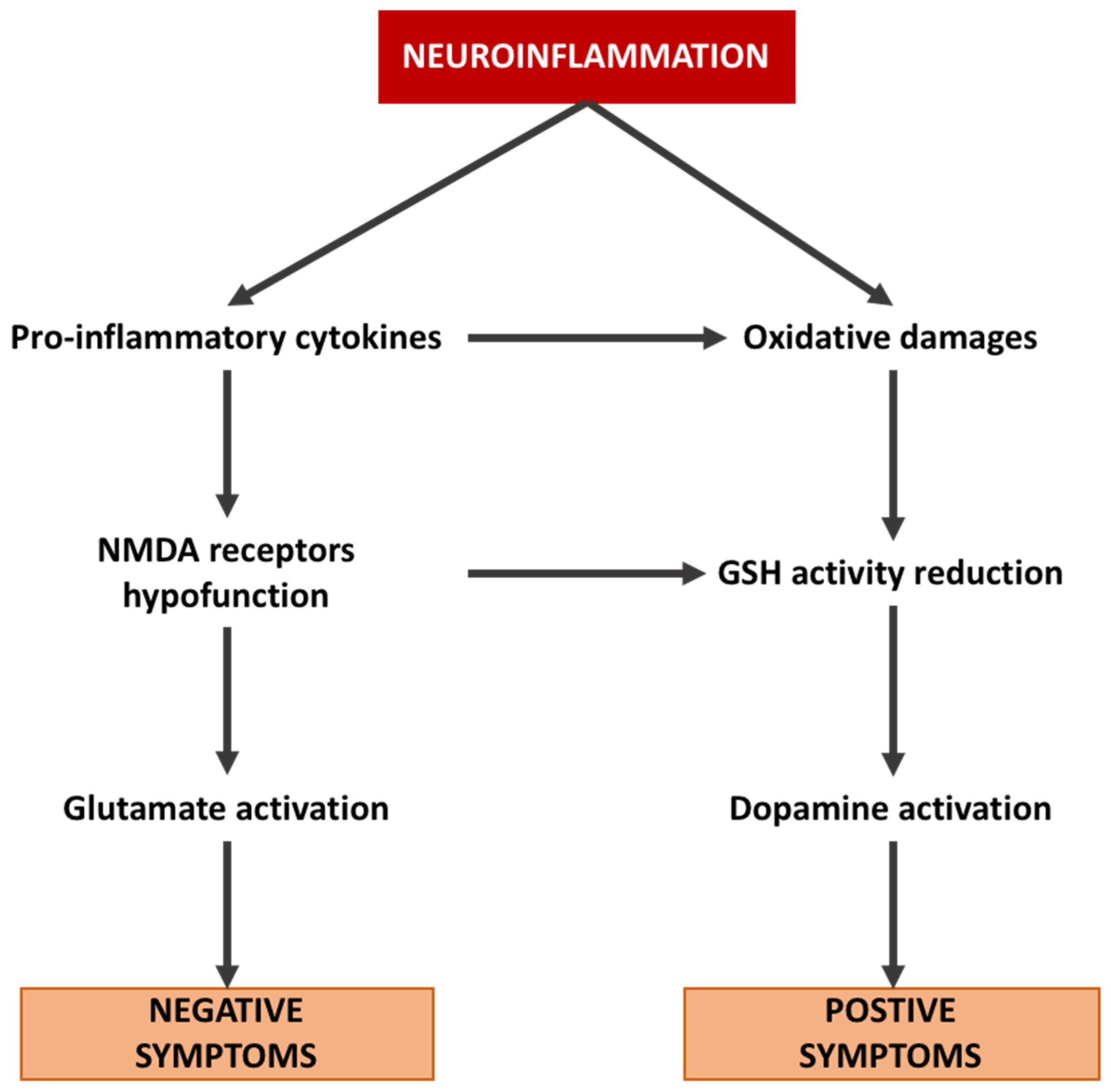
GSH shows a protective role in protecting cells from ROS damage generated by dopamine (DA) metabolism. The observed deficit in GSH results in membrane peroxidation and lesions forming around dopaminergic terminals, leading to a loss of connectivity [114]. Furthermore, DA neurons are controlled by glutamatergic projections to the midbrain DA nuclei [115]. In schizophrenia, DA function is altered by glutamate function [116], while NMDA blockers, such as ketamine, can increase the activation of the DA system [117]. Thus, the NMDA hypofunction observed in schizophrenia may enhance the DA system to be more sensitive to the effects of oxidative stress [118] (Figure 2).
Thus, to corroborate this hypothesis, it turns out that NMDA receptor antagonists, such as ketamine, can lead to psychosis in healthy participants [119]. This psychosis caused by said antagonists reveals symptoms close to those of schizophrenia [120]. Increased GSH levels stimulate the response of NMDA receptors, while the inhibition of GSH leads to decreased NMDA receptor activity [121]. The inhibition of the activity of these receptors is associated with an increase in the production of free radicals and therefore the enhancement of oxidative damage [122]. For this purpose, it should be noted that the synaptic activity of NMDA receptors is mainly related to the production of GSH [123].
7. Interplay between Glutamate and the WNT/β-Catenin Pathway
Physiologically, within astrocytes, β-catenin stimulates the gene expression of EAAT2 and GS [124]. This stimulation is responsible for the reuptake of glutamate from the synaptic cleft by EAAT2 to the astrocytes. Then, GS metabolizes glutamate within these astrocytes.
The β-catenin pathway directly controls the expression of EAAT2, GLT-1, and GS [124,125,126,127]. Thus, the complete inhibition of the β-catenin signaling leads to the cessation of glutamate neurotransmission [125]. The expression of EAAT2 and GS is also modulated by the direct interaction between the β-catenin and its target TCF/LEF [124]. It should be noted that numerous studies have shown the possible role that the NF-κΒ pathway can play in modulating the expression of EAAT2 [126].
8. PPAR α Agonists: Possible Therapeutic Targets in Schizophrenia
Few effective treatments exist for schizophrenia, so it is essential to find new therapeutic pathways. One possible interesting pathway is the use of peroxisome proliferator-activated receptor (PPAR) agonists through their multiple applications in neuroinflammation, oxidative stress, and their interaction with the WNT/β-catenin pathway.
PPARs are ligand-activated transcription factors that bind PPRE (PPAR response elements). PPARs are subdivided into three isoforms: PPARα, PPARγ, and PPAR β/δ [128]. In the nucleus, PPARs form a heterodimer with the retinoid X receptor (RXR) [129]. They are composed of a ligand binding domain that interacts with a DNA binding domain to modulate it [130]. PPARs are involved in several pathophysiological mechanisms, including cell differentiation, protein metabolism, lipid metabolism [131,132], adipocyte differentiation, insulin sensitivity, and inflammation [133,134].
Recent studies have shown that PPARα may modulate the neuroinflammatory mechanisms observed in psychiatric diseases, such as depression [135] and schizophrenia [136]. In schizophrenia, the expression of PPARγ could be increased while PPARα is downregulated, suggesting a metabolic-inflammatory imbalance in its pathogenesis [137]. Note that PPARγ is well known to be expressed inversely to PPARα [138]. The prenatal administration of pioglitazone in a maternal immune rat model, a PPARγ agonist, could attenuate the schizophrenic behavior observed in the male [139]. However, rosiglitazone, another PPARγ agonist, has no significant beneficial effect on the cognitive performance of schizophrenic patients [140]. These contrasting results could be explained by the differences in posology, treatment, and populations. However, stimulating PPARγ would not be the expected therapeutic solution in schizophrenia because of its observed stimulation in schizophrenia.
The decrease in PPARα expression is associated with the development of schizophrenia [141]. It has been shown in numerous studies that the stimulation of PPARα is associated with a decrease in NF-κB pathway activity [142], the downregulation of TNF-α [143], and a decrease in the production of pro-inflammatory cytokines and interferons [144]. Thus, PPARα agonists could play an interesting role in decreasing the neuroinflammation observed in schizophrenia. For this purpose, the stimulation of PPARα increases the brain synthesis of oleoylethanolamide (OEA) and palmitoylethanolamide (PEA) [145]. Both these molecules have anti-inflammatory properties [139]. In addition, one study showed that the decrease in PPARα mRNA levels is concomitant with the increased expression of IL-6 and TNFα [146]. A functional polymorphism L162V in the PPARα gene was observed in a subgroup of schizophrenic patients [147].
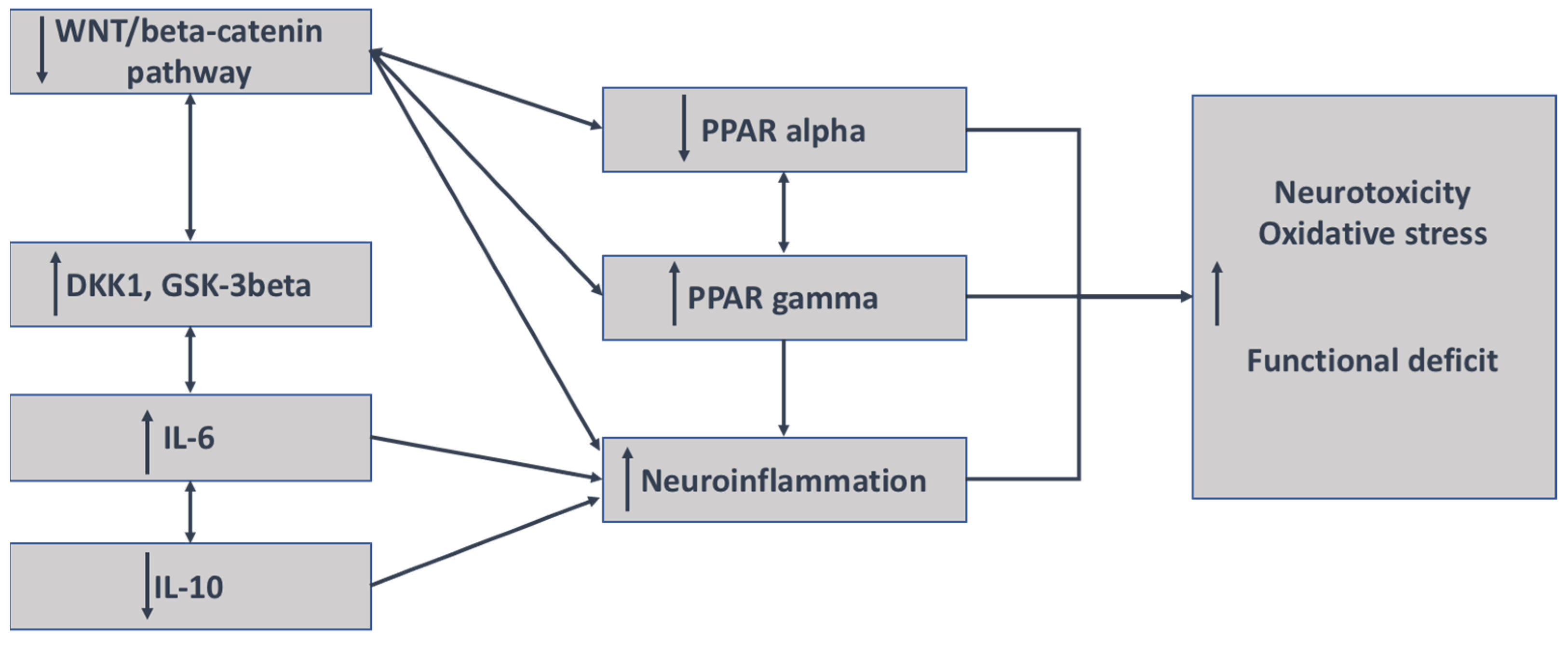
Numerous studies have shown that the WNT/β-catenin pathway and PPARγ act in an opposite manner, including psychiatric disorders, neurodegenerative diseases, and fibrosis processes [80,148,149,150,151,152,153] (Figure 3).
In many diseases, the expression of PPARγ is downregulated by the overexpression of the WNT/β-catenin pathway [154,155,156]. PPARγ is considered a β-catenin target, and when targeted becomes inhibited [157,158]. The WNT/β-catenin pathway and PPARγ interact through a TCF/LEF domain of β-catenin and a catenin binding domain in PPARγ [159,160]. The β-catenin pathway stimulates the PI3K/Akt pathway, leading to a decreased PPARγ expression in 2T2-L1 adipocytes and preadipocytes [161,162]. Recent studies have shown a positive interaction between the WNT/β-catenin pathway and PPARα, elucidating the possible action of this crosstalk to reduce PPARγ expression.
9. Conclusions
A growing body of evidence has shown the important role of neuroinflammation in the pathogenesis of schizophrenia. In addition, the relationship between neuroinflammation and oxidative stress is well documented in this disorder. The WNT/β-catenin pathway is essential for several cellular events that take place in development, homeostasis, and regeneration. However, there is limited knowledge regarding its dysregulation in schizophrenia. Therefore, further studies are still needed in order to clarify the role of the WNT/β-catenin pathway in schizophrenia. Understanding the molecular mechanism of the WNT/β-catenin pathway will contribute to the development of novel therapeutic strategies for schizophrenia. Recently, new evidence has been provided on the protective effects of activation of PPARα expression. In schizophrenia, the WNT/β-catenin pathway and the expression of PPARα act in a positive interplay, which in turn leads to this complex negatively interacting with the expression of PPARγ. This triptych could be a new therapeutic approach to counteract neuroinflammation and oxidative stress in schizophrenia.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ľupták, M.; Michaličková, D.; Fišar, Z.; Kitzlerová, E.; Hroudová, J. Novel Approaches in Schizophrenia-from Risk Factors and Hypotheses to Novel Drug Targets. World J. Psychiatry 2021, 11, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E. Neurodevelopmental Abnormalities in Schizophrenia: Insights from Neuropathology. Dev. Psychopathol. 1999, 11, 439–456. [Google Scholar] [CrossRef]
- Ortiz-Orendain, J.; Castiello-de Obeso, S.; Colunga-Lozano, L.E.; Hu, Y.; Maayan, N.; Adams, C.E. Antipsychotic Combinations for Schizophrenia. Cochrane Database Syst. Rev. 2017, 6, CD009005. [Google Scholar] [CrossRef] [PubMed]
- Heckers, S.; Barch, D.M.; Bustillo, J.; Gaebel, W.; Gur, R.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tandon, R.; Tsuang, M.; et al. Structure of the Psychotic Disorders Classification in DSM-5. Schizophr. Res. 2013, 150, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.E.; Walker, A.K.; Weickert, C.S. Neuroinflammation in Schizophrenia: The Role of Nuclear Factor Kappa B. Transl. Psychiatry 2021, 11, 528. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M. Neuroinflammation and White Matter Pathology in Schizophrenia: Systematic Review. Schizophr. Res. 2015, 161, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Barron, H.; Hafizi, S.; Andreazza, A.C.; Mizrahi, R. Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk. Int. J. Mol. Sci. 2017, 18, 651. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Grainger, S.; Willert, K. Mechanisms of Wnt Signaling and Control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, e1422. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N. Warburg Effect Hypothesis in Autism Spectrum Disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Karabicici, M.; Azbazdar, Y.; Iscan, E.; Ozhan, G. Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases. Membranes 2021, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma. Biomedicines 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Possible Actions of Cannabidiol in Obsessive-Compulsive Disorder by Targeting the WNT/β-Catenin Pathway. Mol. Psychiatry 2021, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jridi, I.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2020, 8, 615131. [Google Scholar] [CrossRef]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The Role of Inflammation in Schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Maes, M.; Berk, M. Schizophrenia Is Primed for an Increased Expression of Depression through Activation of Immuno-Inflammatory, Oxidative and Nitrosative Stress, and Tryptophan Catabolite Pathways. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 101–114. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and Immunity in Schizophrenia: Implications for Pathophysiology and Treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Ilani, T.; Ben-Shachar, D.; Strous, R.D.; Mazor, M.; Sheinkman, A.; Kotler, M.; Fuchs, S. A Peripheral Marker for Schizophrenia: Increased Levels of D3 Dopamine Receptor MRNA in Blood Lymphocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 625–628. [Google Scholar] [CrossRef]
- Kroken, R.A.; Sommer, I.E.; Steen, V.M.; Dieset, I.; Johnsen, E. Constructing the Immune Signature of Schizophrenia for Clinical Use and Research; An Integrative Review Translating Descriptives Into Diagnostics. Front. Psychiatry 2018, 9, 753. [Google Scholar] [CrossRef]
- Rodrigues-Amorim, D.; Rivera-Baltanás, T.; Spuch, C.; Caruncho, H.J.; González-Fernandez, Á.; Olivares, J.M.; Agís-Balboa, R.C. Cytokines Dysregulation in Schizophrenia: A Systematic Review of Psychoneuroimmune Relationship. Schizophr. Res. 2018, 197, 19–33. [Google Scholar] [CrossRef]
- Boerrigter, D.; Weickert, T.W.; Lenroot, R.; O’Donnell, M.; Galletly, C.; Liu, D.; Burgess, M.; Cadiz, R.; Jacomb, I.; Catts, V.S.; et al. Using Blood Cytokine Measures to Define High Inflammatory Biotype of Schizophrenia and Schizoaffective Disorder. J. Neuroinflammation 2017, 14, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osimo, E.F.; Cardinal, R.N.; Jones, P.B.; Khandaker, G.M. Prevalence and Correlates of Low-Grade Systemic Inflammation in Adult Psychiatric Inpatients: An Electronic Health Record-Based Study. Psychoneuroendocrinology 2018, 91, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; Lançon, C.; Korchia, T.; Auquier, P.; Boyer, L. The Role of Inflammation in the Treatment of Schizophrenia. Front. Psychiatry 2020, 11, 160. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A Meta-Analysis of Blood Cytokine Network Alterations in Psychiatric Patients: Comparisons between Schizophrenia, Bipolar Disorder and Depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.K.; Miller, B.J. Meta-Analysis of Cerebrospinal Fluid Cytokine and Tryptophan Catabolite Alterations in Psychiatric Patients: Comparisons Between Schizophrenia, Bipolar Disorder, and Depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Herberth, M.; Rahmoune, H.; Schwarz, E.; Koethe, D.; Harris, L.W.; Kranaster, L.; Witt, S.H.; Spain, M.; Barnes, A.; Schmolz, M.; et al. Identification of a Molecular Profile Associated with Immune Status in First-Onset Schizophrenia Patients. Clin. Schizophr. Relat. Psychoses 2014, 7, 207–215. [Google Scholar] [CrossRef]
- Miller, B.J.; Gassama, B.; Sebastian, D.; Buckley, P.; Mellor, A. Meta-Analysis of Lymphocytes in Schizophrenia: Clinical Status and Antipsychotic Effects. Biol. Psychiatry 2013, 73, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, P.; Chi, D.; Wu, T.; Mei, Z.; Cui, G. Association between C-Reactive Protein and Risk of Schizophrenia: An Updated Meta-Analysis. Oncotarget 2017, 8, 75445–75454. [Google Scholar] [CrossRef] [Green Version]
- Liemburg, E.J.; Nolte, I.M.; PHAMOUS Investigators; Klein, H.C.; Knegtering, H. Relation of Inflammatory Markers with Symptoms of Psychotic Disorders: A Large Cohort Study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 89–94. [Google Scholar] [CrossRef]
- Fathian, F.; Løberg, E.-M.; Gjestad, R.; Steen, V.M.; Kroken, R.A.; Jørgensen, H.A.; Johnsen, E. Associations between C-Reactive Protein Levels and Cognition during the First 6 Months after Acute Psychosis. Acta Neuropsychiatr. 2019, 31, 36–45. [Google Scholar] [CrossRef]
- Fond, G.; Godin, O.; Boyer, L.; Berna, F.; Andrianarisoa, M.; Coulon, N.; Brunel, L.; Bulzacka, E.; Aouizerate, B.; Capdevielle, D.; et al. Chronic Low-Grade Peripheral Inflammation Is Associated with Ultra Resistant Schizophrenia. Results from the FACE-SZ Cohort. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderholm, K.R.; Skogh, E.; Olsson, S.K.; Dahl, M.-L.; Holtze, M.; Engberg, G.; Samuelsson, M.; Erhardt, S. Increased Levels of Kynurenine and Kynurenic Acid in the CSF of Patients with Schizophrenia. Schizophr. Bull. 2012, 38, 426–432. [Google Scholar] [CrossRef]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. PPARγ Agonists: Potential Treatment for Autism Spectrum Disorder by Inhibiting the Canonical WNT/β-Catenin Pathway. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Al-Harthi, L. Wnt/β-Catenin and Its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt Your Brain Be Inflamed? Yes, It Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome Proliferator-Activated Receptor Gamma Activation Can Regulate Beta-Catenin Levels via a Proteasome-Mediated and Adenomatous Polyposis Coli-Independent Pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 Expression in Transgenic Mouse Models of Neurodegenerative Disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Effects of Cannabidiol Interactions with Wnt/β-Catenin Pathway and PPARγ on Oxidative Stress and Neuroinflammation in Alzheimer’s Disease. Acta Biochim. Biophys. Sin. 2017, 49, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt Signaling in the Nervous System and in Alzheimer’s Disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.H.; Mors, O.; Mortensen, P.B.; et al. Common Variants Conferring Risk of Schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okerlund, N.D.; Cheyette, B.N.R. Synaptic Wnt Signaling-a Contributor to Major Psychiatric Disorders? J. Neurodev. Disord. 2011, 3, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.-G.; Steiner, J.; Bogerts, B.; Braun, K.; Braun, A.K.; Jankowski, Z.; et al. The Role of Dopamine in Schizophrenia from a Neurobiological and Evolutionary Perspective: Old Fashioned, but Still in Vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, D.; Kerwin, R.; al-Sarraji, S.; Brion, J.P.; Chadwich, A.; Lovestone, S.; Anderton, B.; Everall, I. Abnormalities of Wnt Signalling in Schizophrenia--Evidence for Neurodevelopmental Abnormality. Neuroreport 1998, 9, 1379–1383. [Google Scholar] [CrossRef]
- Alsabban, A.H.; Morikawa, M.; Tanaka, Y.; Takei, Y.; Hirokawa, N. Kinesin Kif3b Mutation Reduces NMDAR Subunit NR2A Trafficking and Causes Schizophrenia-like Phenotypes in Mice. EMBO J. 2020, 39, e101090. [Google Scholar] [CrossRef]
- Lovestone, S.; Killick, R.; Di Forti, M.; Murray, R. Schizophrenia as a GSK-3 Dysregulation Disorder. Trends Neurosci. 2007, 30, 142–149. [Google Scholar] [CrossRef]
- Ftouh, S.; Akbar, M.T.; Hirsch, S.R.; de Belleroche, J.S. Down-Regulation of Dickkopf 3, a Regulator of the Wnt Signalling Pathway, in Elderly Schizophrenic Subjects. J. Neurochem. 2005, 94, 520–530. [Google Scholar] [CrossRef]
- Proitsi, P.; Li, T.; Hamilton, G.; Di Forti, M.; Collier, D.; Killick, R.; Chen, R.; Sham, P.; Murray, R.; Powell, J.; et al. Positional Pathway Screen of Wnt Signaling Genes in Schizophrenia: Association with DKK4. Biol. Psychiatry 2008, 63, 13–16. [Google Scholar] [CrossRef]
- Hoseth, E.Z.; Krull, F.; Dieset, I.; Mørch, R.H.; Hope, S.; Gardsjord, E.S.; Steen, N.E.; Melle, I.; Brattbakk, H.-R.; Steen, V.M.; et al. Exploring the Wnt Signaling Pathway in Schizophrenia and Bipolar Disorder. Transl. Psychiatry 2018, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Low, S.-K.; Atkins, J.R.; Wu, J.Q.; Reay, W.R.; Cairns, H.M.; Green, M.J.; Schall, U.; Jablensky, A.; Mowry, B.; et al. Wnt Receptor Gene FZD1 Was Associated with Schizophrenia in Genome-Wide SNP Analysis of the Australian Schizophrenia Research Bank Cohort. Aust. N. Z. J. Psychiatry 2020, 54, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Katsu, T.; Ujike, H.; Nakano, T.; Tanaka, Y.; Nomura, A.; Nakata, K.; Takaki, M.; Sakai, A.; Uchida, N.; Imamura, T.; et al. The Human Frizzled-3 (FZD3) Gene on Chromosome 8p21, a Receptor Gene for Wnt Ligands, Is Associated with the Susceptibility to Schizophrenia. Neurosci. Lett. 2003, 353, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Si, T.; Ling, Y.; Ruan, Y.; Han, Y.; Wang, X.; Zhang, H.; Kong, Q.; Li, X.; Liu, C.; et al. Association Study of the Human FZD3 Locus with Schizophrenia. Biol. Psychiatry 2003, 54, 1298–1301. [Google Scholar] [CrossRef]
- Ide, M.; Muratake, T.; Yamada, K.; Iwayama-Shigeno, Y.; Iwamoto, K.; Takao, H.; Toyota, T.; Kaneko, N.; Minabe, Y.; Nakamura, K.; et al. Genetic and Expression Analyses of FZD3 in Schizophrenia. Biol. Psychiatry 2004, 56, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-KappaB by Toll-like Receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S. Innate Immunity and Neuroinflammation in the CNS: The Role of Microglia in Toll-like Receptor-Mediated Neuronal Injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pinilla, M.; Rodriguez-Peralto, J.L.; Hitt, R.; Sanchez, J.J.; Sanchez-Verde, L.; Alameda, F.; Ballestin, C.; Sanchez-Cespedes, M. Beta-Catenin, Nf-KappaB and FAS Protein Expression Are Independent Events in Head and Neck Cancer: Study of Their Association with Clinical Parameters. Cancer Lett. 2005, 230, 141–148. [Google Scholar] [CrossRef]
- Sun, J.; Hobert, M.E.; Duan, Y.; Rao, A.S.; He, T.-C.; Chang, E.B.; Madara, J.L. Crosstalk between NF-KappaB and Beta-Catenin Pathways in Bacterial-Colonized Intestinal Epithelial Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G129–G137. [Google Scholar] [CrossRef]
- Moreau, M.; Mourah, S.; Dosquet, C. β-Catenin and NF-ΚB Cooperate to Regulate the UPA/UPAR System in Cancer Cells. Int. J. Cancer 2011, 128, 1280–1292. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-Ubiquitin Ligase Complex Associates Specifically with Phosphorylated Destruction Motifs in IkappaBalpha and Beta-Catenin and Stimulates IkappaBalpha Ubiquitination in Vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-KappaB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and Adaptive Immune Responses Regulated by Glycogen Synthase Kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Liao, A.P.; Kuppireddi, S.; Ye, Z.; Ciancio, M.J.; Sun, J. Beta-Catenin Activity Negatively Regulates Bacteria-Induced Inflammation. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Miller, S.A.; Wang, H.-Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.-Y.; Hung, M.-C. Beta-Catenin Interacts with and Inhibits NF-Kappa B in Human Colon and Breast Cancer. Cancer Cell 2002, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liao, Y.; Ma, K.; Wang, Y.; Zhang, G.; Yang, R.; Deng, J. PI3K Is Required for the Physical Interaction and Functional Inhibition of NF-ΚB by β-Catenin in Colorectal Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 434, 760–766. [Google Scholar] [CrossRef]
- Giacoppo, S.; Soundara Rajan, T.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. Moringin Activates Wnt Canonical Pathway by Inhibiting GSK3β in a Mouse Model of Experimental Autoimmune Encephalomyelitis. Drug Des. Devel. Ther. 2016, 10, 3291–3304. [Google Scholar] [CrossRef] [Green Version]
- De Ferrari, G.V.; Avila, M.E.; Medina, M.A.; Perez-Palma, E.; Bustos, B.I.; Alarcon, M.A. Wnt/β-Catenin Signaling in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2014, 13, 745–754. [Google Scholar] [CrossRef]
- Le Grand, J.N.; Gonzalez-Cano, L.; Pavlou, M.A.; Schwamborn, J.C. Neural Stem Cells in Parkinson’s Disease: A Role for Neurogenesis Defects in Onset and Progression. Cell. Mol. Life Sci. CMLS 2015, 72, 773–797. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int. J. Mol. Sci. 2017, 18, 2537. [Google Scholar] [CrossRef] [Green Version]
- Roomruangwong, C.; Noto, C.; Kanchanatawan, B.; Anderson, G.; Kubera, M.; Carvalho, A.F.; Maes, M. The Role of Aberrations in the Immune-Inflammatory Response System (IRS) and the Compensatory Immune-Regulatory Reflex System (CIRS) in Different Phenotypes of Schizophrenia: The IRS-CIRS Theory of Schizophrenia. Mol. Neurobiol. 2020, 57, 778–797. [Google Scholar] [CrossRef] [PubMed]
- Al-Dujaili, A.H.; Mousa, R.F.; Al-Hakeim, H.K.; Maes, M. High Mobility Group Protein 1 and Dickkopf-Related Protein 1 in Schizophrenia and Treatment-Resistant Schizophrenia: Associations With Interleukin-6, Symptom Domains, and Neurocognitive Impairments. Schizophr. Bull. 2021, 47, 530–541. [Google Scholar] [CrossRef]
- Maes, M.; Sirivichayakul, S.; Kanchanatawan, B.; Carvalho, A.F. In Schizophrenia, Psychomotor Retardation Is Associated with Executive and Memory Impairments, Negative and Psychotic Symptoms, Neurotoxic Immune Products and Lower Natural IgM to Malondialdehyde. World J. Biol. Psychiatry Off. J. World Fed. Soc. Biol. Psychiatry 2020, 21, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Vojdani, A.; Sirivichayakul, S.; Barbosa, D.S.; Kanchanatawan, B. Inflammatory and Oxidative Pathways Are New Drug Targets in Multiple Episode Schizophrenia and Leaky Gut, Klebsiella Pneumoniae, and C1q Immune Complexes Are Additional Drug Targets in First Episode Schizophrenia. Mol. Neurobiol. 2021, 58, 3319–3334. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-L.; Yang, C.-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. BioMed Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef]
- Turillazzi, E.; Neri, M.; Cerretani, D.; Cantatore, S.; Frati, P.; Moltoni, L.; Busardò, F.P.; Pomara, C.; Riezzo, I.; Fineschi, V. Lipid Peroxidation and Apoptotic Response in Rat Brain Areas Induced by Long-Term Administration of Nandrolone: The Mutual Crosstalk between ROS and NF-KB. J. Cell. Mol. Med. 2016, 20, 601–612. [Google Scholar] [CrossRef]
- Conus, P.; Seidman, L.J.; Fournier, M.; Xin, L.; Cleusix, M.; Baumann, P.S.; Ferrari, C.; Cousins, A.; Alameda, L.; Gholam-Rezaee, M.; et al. N-Acetylcysteine in a Double-Blind Randomized Placebo-Controlled Trial: Toward Biomarker-Guided Treatment in Early Psychosis. Schizophr. Bull. 2018, 44, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Sommer, I.E.; Bearden, C.E.; van Dellen, E.; Breetvelt, E.J.; Duijff, S.N.; Maijer, K.; van Amelsvoort, T.; de Haan, L.; Gur, R.E.; Arango, C.; et al. Early Interventions in Risk Groups for Schizophrenia: What Are We Waiting For? NPJ Schizophr. 2016, 2, 16003. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Pearson, R.M.; Zammit, S.; Lewis, G.; Jones, P.B. Association of Serum Interleukin 6 and C-Reactive Protein in Childhood with Depression and Psychosis in Young Adult Life: A Population-Based Longitudinal Study. JAMA Psychiatry 2014, 71, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Pedraz-Petrozzi, B.; Elyamany, O.; Rummel, C.; Mulert, C. Effects of Inflammation on the Kynurenine Pathway in Schizophrenia - a Systematic Review. J. Neuroinflammation 2020, 17, 56. [Google Scholar] [CrossRef]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The Role of Oxidative Stress during Inflammatory Processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordt, E.A.; Polster, B.M. NADPH Oxidase- and Mitochondria-Derived Reactive Oxygen Species in Proinflammatory Microglial Activation: A Bipartisan Affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, M.; Serritella, A.V.; Sawa, A.; Sedlak, T.W. Implications for Reactive Oxygen Species in Schizophrenia Pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.Y.; Woo, T.-U.W. Oxidative Stress in Schizophrenia: An Integrated Approach. Neurosci. Biobehav. Rev. 2011, 35, 878–893. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Yao, J.K.; Reddy, R.D.; van Kammen, D.P. Oxidative Damage and Schizophrenia: An Overview of the Evidence and Its Therapeutic Implications. CNS Drugs 2001, 15, 287–310. [Google Scholar] [CrossRef]
- Khan, M.M.; Evans, D.R.; Gunna, V.; Scheffer, R.E.; Parikh, V.V.; Mahadik, S.P. Reduced Erythrocyte Membrane Essential Fatty Acids and Increased Lipid Peroxides in Schizophrenia at the Never-Medicated First-Episode of Psychosis and after Years of Treatment with Antipsychotics. Schizophr. Res. 2002, 58, 1–10. [Google Scholar] [CrossRef]
- Raffa, M.; Atig, F.; Mhalla, A.; Kerkeni, A.; Mechri, A. Decreased Glutathione Levels and Impaired Antioxidant Enzyme Activities in Drug-Naive First-Episode Schizophrenic Patients. BMC Psychiatry 2011, 11, 124. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; Zheng, Y.L.; Xiu, M.H.; Chen, D.C.; Kosten, T.R.; Zhang, X.Y. Reduced Plasma Total Antioxidant Status in First-Episode Drug-Naive Patients with Schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1064–1067. [Google Scholar] [CrossRef]
- Miyaoka, T.; Ieda, M.; Hashioka, S.; Wake, R.; Furuya, M.; Liaury, K.; Hayashida, M.; Tsuchie, K.; Arauchi, R.; Araki, T.; et al. Analysis of Oxidative Stress Expressed by Urinary Level of Biopyrrins and 8-Hydroxydeoxyguanosine in Patients with Chronic Schizophrenia. Psychiatry Clin. Neurosci. 2015, 69, 693–698. [Google Scholar] [CrossRef]
- Dietrich-Muszalska, A.; Kwiatkowska, A. Generation of Superoxide Anion Radicals and Platelet Glutathione Peroxidase Activity in Patients with Schizophrenia. Neuropsychiatr. Dis. Treat. 2014, 10, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solberg, D.K.; Refsum, H.; Andreassen, O.A.; Bentsen, H. A Five-Year Follow-up Study of Antioxidants, Oxidative Stress and Polyunsaturated Fatty Acids in Schizophrenia. Acta Neuropsychiatr. 2019, 31, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.K.; Leonard, S.; Reddy, R. Altered Glutathione Redox State in Schizophrenia. Dis. Markers 2006, 22, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawryluk, J.W.; Wang, J.-F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased Levels of Glutathione, the Major Brain Antioxidant, in Post-Mortem Prefrontal Cortex from Patients with Psychiatric Disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.; Dedova, I.; Cordwell, S.; Matsumoto, I. A Proteome Analysis of the Anterior Cingulate Cortex Gray Matter in Schizophrenia. Mol. Psychiatry 2006, 11, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Fraguas, D.; Gonzalez-Pinto, A.; Micó, J.A.; Reig, S.; Parellada, M.; Martínez-Cengotitabengoa, M.; Castro-Fornieles, J.; Rapado-Castro, M.; Baeza, I.; Janssen, J.; et al. Decreased Glutathione Levels Predict Loss of Brain Volume in Children and Adolescents with First-Episode Psychosis in a Two-Year Longitudinal Study. Schizophr. Res. 2012, 137, 58–65. [Google Scholar] [CrossRef]
- Lavoie, S.; Murray, M.M.; Deppen, P.; Knyazeva, M.G.; Berk, M.; Boulat, O.; Bovet, P.; Bush, A.I.; Conus, P.; Copolov, D.; et al. Glutathione Precursor, N-Acetyl-Cysteine, Improves Mismatch Negativity in Schizophrenia Patients. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Bentsen, H.; Landrø, N.I. Neurocognitive Effects of an Omega-3 Fatty Acid and Vitamins E+C in Schizophrenia: A Randomised Controlled Trial. Prostaglandins Leukot. Essent. Fatty Acids 2018, 136, 57–66. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and Psychological Disorders. Curr. Neuropharmacol. 2014, 12, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Lin, C.-L.G.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate Transporter EAAT2: A New Target for the Treatment of Neurodegenerative Diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Kornhuber, H.H.; Schmid-Burgk, W.; Holzmüller, B. Low Cerebrospinal Fluid Glutamate in Schizophrenic Patients and a New Hypothesis on Schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Hashimoto, K. Targeting of NMDA Receptors in New Treatments for Schizophrenia. Expert Opin. Ther. Targets 2014, 18, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Grima, G.; Benz, B.; Parpura, V.; Cuénod, M.; Do, K.Q. Dopamine-Induced Oxidative Stress in Neurons with Glutathione Deficit: Implication for Schizophrenia. Schizophr. Res. 2003, 62, 213–224. [Google Scholar] [CrossRef]
- Miller, D.W.; Abercrombie, E.D. Effects of MK-801 on Spontaneous and Amphetamine-Stimulated Dopamine Release in Striatum Measured with in Vivo Microdialysis in Awake Rats. Brain Res. Bull. 1996, 40, 57–62. [Google Scholar] [CrossRef]
- McGuire, P.K.; Bench, C.J.; Frith, C.D.; Marks, I.M.; Frackowiak, R.S.; Dolan, R.J. Functional Anatomy of Obsessive-Compulsive Phenomena. Br. J. Psychiatry J. Ment. Sci. 1994, 164, 459–468. [Google Scholar] [CrossRef]
- Jackson, M.E.; Homayoun, H.; Moghaddam, B. NMDA Receptor Hypofunction Produces Concomitant Firing Rate Potentiation and Burst Activity Reduction in the Prefrontal Cortex. Proc. Natl. Acad. Sci. USA 2004, 101, 8467–8472. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and Dopamine in Schizophrenia: An Update for the 21st Century. J. Psychopharmacol. Oxf. Engl. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-H.; Lane, H.-Y. Early Identification and Intervention of Schizophrenia: Insight From Hypotheses of Glutamate Dysfunction and Oxidative Stress. Front. Psychiatry 2019, 10, 93. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic Effects of the Noncompetitive NMDA Antagonist, Ketamine, in Humans. Psychotomimetic, Perceptual, Cognitive, and Neuroendocrine Responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K.Q. Redox Dysregulation, Neuroinflammation, and NMDA Receptor Hypofunction: A “Central Hub” in Schizophrenia Pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadia, S.; Soriano, F.X.; Léveillé, F.; Martel, M.-A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA Receptor Activity Boosts Intrinsic Antioxidant Defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA Receptor Activity Is Coupled to the Transcriptional Control of the Glutathione System. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgen, V.; Narasipura, S.D.; Sharma, A.; Min, S.; Al-Harthi, L. β-Catenin Signaling Positively Regulates Glutamate Uptake and Metabolism in Astrocytes. J. Neuroinflammation 2016, 13, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasipura, S.D.; Henderson, L.J.; Fu, S.W.; Chen, L.; Kashanchi, F.; Al-Harthi, L. Role of β-Catenin and TCF/LEF Family Members in Transcriptional Activity of HIV in Astrocytes. J. Virol. 2012, 86, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Eid, T.; Tu, N.; Lee, T.-S.W.; Lai, J.C.K. Regulation of Astrocyte Glutamine Synthetase in Epilepsy. Neurochem. Int. 2013, 63, 670–681. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Henderson, L.J.; Major, E.O.; Al-Harthi, L. IFN-Gamma Mediates Enhancement of HIV Replication in Astrocytes by Inducing an Antagonist of the Beta-Catenin Pathway (DKK1) in a STAT 3-Dependent Manner. J. Immunol. Baltim. 2011, 186, 6771–6778. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.D.; Plutzky, J. Peroxisome Proliferator-Activated Receptors as Transcriptional Nodal Points and Therapeutic Targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef]
- Smirnov, A.N. Nuclear Receptors: Nomenclature, Ligands, Mechanisms of Their Effects on Gene Expression. Biochem. Biokhimiia 2002, 67, 957–977. [Google Scholar] [CrossRef]
- Kota, B.P.; Huang, T.H.-W.; Roufogalis, B.D. An Overview on Biological Mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef]
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, N.; Duez, H.; Fruchart, J.-C.; Staels, B. Peroxisome Proliferator-Activated Receptors and Atherogenesis: Regulators of Gene Expression in Vascular Cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Cunard, R.; Ricote, M.; DiCampli, D.; Archer, D.C.; Kahn, D.A.; Glass, C.K.; Kelly, C.J. Regulation of Cytokine Expression by Ligands of Peroxisome Proliferator Activated Receptors. J. Immunol. 2002, 168, 2795–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and Molecular Mechanisms of Transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Wang, Y.-J.; Wang, H.; Song, L.; Huang, C.; Zhu, Q.; Wu, F.; Zhang, W. Antidepressant-like Effects of Fenofibrate in Mice via the Hippocampal Brain-Derived Neurotrophic Factor Signalling Pathway. Br. J. Pharmacol. 2017, 174, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Rolland, B.; Deguil, J.; Jardri, R.; Cottencin, O.; Thomas, P.; Bordet, R. Therapeutic Prospects of PPARs in Psychiatric Disorders: A Comprehensive Review. Curr. Drug Targets 2013, 14, 724–732. [Google Scholar] [CrossRef]
- Chase, K.A.; Rosen, C.; Gin, H.; Bjorkquist, O.; Feiner, B.; Marvin, R.; Conrin, S.; Sharma, R.P. Metabolic and Inflammatory Genes in Schizophrenia. Psychiatry Res. 2015, 225, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- De Felice, M.; Melis, M.; Aroni, S.; Muntoni, A.L.; Fanni, S.; Frau, R.; Devoto, P.; Pistis, M. The PPARα Agonist Fenofibrate Attenuates Disruption of Dopamine Function in a Maternal Immune Activation Rat Model of Schizophrenia. CNS Neurosci. Ther. 2019, 25, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Fan, X.; Wang, J.; Liu, D.; Freudenreich, O.; Goff, D.; Henderson, D.C. Rosiglitazone and Cognitive Function in Clozapine-Treated Patients with Schizophrenia: A Pilot Study. Psychiatry Res. 2012, 200, 79–82. [Google Scholar] [CrossRef]
- Costa, M.; Squassina, A.; Congiu, D.; Chillotti, C.; Niola, P.; Galderisi, S.; Pistis, M.; Del Zompo, M. Investigation of Endocannabinoid System Genes Suggests Association between Peroxisome Proliferator Activator Receptor-α Gene (PPARA) and Schizophrenia. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2013, 23, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.-C.; Robbins, M.E. PPARalpha Ligands Inhibit Radiation-Induced Microglial Inflammatory Responses by Negatively Regulating NF-KappaB and AP-1 Pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.R.; Clarke, S.; Rodgers, K.; Thornhill, B.; Peters, J.M.; Gonzalez, F.J.; Gimble, J.M. Effect of Peroxisome Proliferator-Activated Receptor Alpha Activators on Tumor Necrosis Factor Expression in Mice during Endotoxemia. Infect. Immun. 1999, 67, 3488–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-M.; Chu, K.-M.; Lai, J.-H. The Modulatory Mechanisms of Fenofibrate on Human Primary T Cells. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2010, 40, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Carta, G.; Pistis, M.; Banni, S. Physiological Role of Peroxisome Proliferator-Activated Receptors Type α on Dopamine Systems. CNS Neurol. Disord. Drug Targets 2013, 12, 70–77. [Google Scholar] [CrossRef]
- Sagheddu, C.; Melis, M.; Muntoni, A.L.; Pistis, M. Repurposing Peroxisome Proliferator-Activated Receptor Agonists in Neurological and Psychiatric Disorders. Pharm. Basel Switz. 2021, 14, 1025. [Google Scholar] [CrossRef]
- Nadalin, S.; Giacometti, J.; Buretić-Tomljanović, A. PPARα-L162V Polymorphism Is Not Associated with Schizophrenia Risk in a Croatian Population. Prostaglandins Leukot. Essent. Fatty Acids 2014, 91, 221–225. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-Β1, Canonical WNT/β-Catenin Pathway and PPAR γ in Radiation-Induced Fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromolecular Med. 2018, 20, 174–204. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Guillevin, R.; Lecarpentier, Y. Interactions Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma on Neuroinflammation, Demyelination, and Remyelination in Multiple Sclerosis. Cell. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay Between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Hypothesis of Opposite Interplay Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, M.; Rahman, N.; Kim, Y.-S. Wnt/β-Catenin Signaling Plays a Distinct Role in Methyl Gallate-Mediated Inhibition of Adipogenesis. Biochem. Biophys. Res. Commun. 2016, 479, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Niu, M.; Zhai, X.; Zhou, Q.; Zhou, Y. β-Catenin Pathway Is Required for TGF-Β1 Inhibition of PPARγ Expression in Cultured Hepatic Stellate Cells. Pharmacol. Res. 2012, 66, 219–225. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, S.H.; Lee, Y.J.; Kang, E.S.; Lee, B.-W.; Cha, B.S.; Kim, J.W.; Song, D.H.; Lee, H.C. Transcription Factor Snail Is a Novel Regulator of Adipocyte Differentiation via Inhibiting the Expression of Peroxisome Proliferator-Activated Receptor γ. Cell. Mol. Life Sci. CMLS 2013, 70, 3959–3971. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; D’Urso, M.C.; di Blasio, G.; Brignone, M.S.; De Simone, R.; Minghetti, L. Glycogen Synthase Kinase 3 Is Part of the Molecular Machinery Regulating the Adaptive Response to LPS Stimulation in Microglial Cells. Brain. Behav. Immun. 2016, 55, 225–235. [Google Scholar] [CrossRef]
- Jansson, E.A.; Are, A.; Greicius, G.; Kuo, I.-C.; Kelly, D.; Arulampalam, V.; Pettersson, S. The Wnt/Beta-Catenin Signaling Pathway Targets PPARgamma Activity in Colon Cancer Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Zuo, Y.; Farmer, S.R. Functional Interaction between Peroxisome Proliferator-Activated Receptor Gamma and Beta-Catenin. Mol. Cell. Biol. 2006, 26, 5827–5837. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Carson, D.A. Repression of Beta-Catenin Signaling by PPAR Gamma Ligands. Eur. J. Pharmacol. 2010, 636, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Moldes, M.; Zuo, Y.; Morrison, R.F.; Silva, D.; Park, B.-H.; Liu, J.; Farmer, S.R. Peroxisome-Proliferator-Activated Receptor Gamma Suppresses Wnt/Beta-Catenin Signalling during Adipogenesis. Biochem. J. 2003, 376, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsken, J.; Behrens, J. The Wnt Signalling Pathway. J. Cell Sci. 2002, 115, 3977–3978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of the mechanism for the interaction between neuroinflammation and oxidative stress in schizophrenia. A vicious circle can occur in which these processes stimulate each other, leading to psychotic symptoms.
Figure 1.
Schematic representation of the mechanism for the interaction between neuroinflammation and oxidative stress in schizophrenia. A vicious circle can occur in which these processes stimulate each other, leading to psychotic symptoms.

Figure 2.
Interaction between neuroinflammation, glutamate pathway, dopamine pathway, and subsequent stimulation of positive and negative symptoms in schizophrenia.
Figure 2.
Interaction between neuroinflammation, glutamate pathway, dopamine pathway, and subsequent stimulation of positive and negative symptoms in schizophrenia.

Figure 3.
Mechanisms of interaction between neuroinflammation and the WNT/β-catenin pathway in schizophrenia. The decrease in the WNT/β-catenin pathway is associated with the increase in both DKK1 and GSK-3 β and their inhibitors; the stimulation of IL-6 and IL-8 expression; and a decrease in the expression of IL-10, an antagonist marker of inflammation. The decrease in WNT/β-catenin leads to the upregulation of PPARγ but the downregulation of PPAR α. An increase in the expression of PPARγ leads to the involvement of neuroinflammation.
Figure 3.
Mechanisms of interaction between neuroinflammation and the WNT/β-catenin pathway in schizophrenia. The decrease in the WNT/β-catenin pathway is associated with the increase in both DKK1 and GSK-3 β and their inhibitors; the stimulation of IL-6 and IL-8 expression; and a decrease in the expression of IL-10, an antagonist marker of inflammation. The decrease in WNT/β-catenin leads to the upregulation of PPARγ but the downregulation of PPAR α. An increase in the expression of PPARγ leads to the involvement of neuroinflammation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vallée, A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway. Int. J. Mol. Sci. 2022, 23, 2810. https://doi.org/10.3390/ijms23052810
AMA Style
Vallée A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway. International Journal of Molecular Sciences. 2022; 23(5):2810. https://doi.org/10.3390/ijms23052810
Chicago/Turabian StyleVallée, Alexandre. 2022. "Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway" International Journal of Molecular Sciences 23, no. 5: 2810. https://doi.org/10.3390/ijms23052810
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.