
Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease


1
Instituto de Biomedicina de Valencia (CSIC), Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Jaime Roig 11, 46010 Valencia, Spain
2
IIS Fundación Jiménez Díaz, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), 28040 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(10), 5351; https://doi.org/10.3390/ijms22105351
Submission received: 20 April 2021
/
Revised: 12 May 2021
/
Accepted: 17 May 2021
/
Published: 19 May 2021
(This article belongs to the Special Issue Tissue-Specific Direct and Indirect Effects of Metformin and Its Application)
{kind=link}
{kind=link}
Abstract
:Metformin is a drug in the family of biguanide compounds that is widely used in the treatment of type 2 diabetes (T2D). Interestingly, the therapeutic potential of metformin expands its prescribed use as an anti-diabetic drug. In this sense, it has been described that metformin administration has beneficial effects on different neurological conditions. In this work, we review the beneficial effects of this drug as a neuroprotective agent in different neurological diseases, with a special focus on epileptic disorders and Lafora disease, a particular type of progressive myoclonus epilepsy. In addition, we review the different proposed mechanisms of action of metformin to understand its function at the neurological level.
Keywords:
AMPK; epilepsy; GPD2; Lafora disease; mechanism of action; metformin; neurological disorders1. Introduction
Metformin (N,N-Dimethylimidodicarbonimidic diamide) belongs to the family of biguanide compounds that have glucose-lowering effects. Among the biguanide family of compounds, metformin was initially of little clinical interest, due to its low potency requiring high doses of the compound to be effective. However, metformin showed a higher safety profile than its counterparts, such as phenformin or buformin, which were discarded for clinical use because they produced lactic acidosis (reviewed in [1]). Metformin is currently the most commonly prescribed drug for type 2 diabetes (T2D), and is taken by an estimated 150 million people worldwide. It has the advantage over other non-insulin-based diabetes therapies of reducing blood glucose levels without inducing hypoglycemia. Due to its superior safety profile, it has become the first-line treatment for T2D, and is now featured on the World Health Organization’s essential medicines list [1,2,3]. However, although metformin is usually well tolerated, it does have some side effects. In some patients, it can produce lactic acidosis, gastrointestinal discomfort, and vitamin B12 deficiency. For this reason, metformin should be administered initially at low doses that are increased if side effects do not appear [4].
The regular dose of metformin used in diabetic patients is 1–2 g/day, leading to a plasma metformin concentration of 50–100 μM. Numerous reports on the possible metformin mechanisms of action have been published, and some contradictory results have been shown due to differences in the cellular system used in the analyses, or because different doses of metformin were used (reviewed in [1]). Thus, different mechanisms of action have been described depending on the dose used in rats and mice: (1) supra-pharmacological metformin doses (>250 mg/kg/day) achieved >1 mM plasma metformin concentration; and (2) regular pharmacological treatment (50–100 mg/kg/day) achieved 50–100 μM metformin concentration, which corresponds to the regular human dose mentioned above [1,2,3]. Thus, depending on the dose, the mechanism of action of metformin can be explained by it affecting different signaling pathways (see below).
According to PubChem [PubChem. Available online: http://pubchem.ncbi.nlm.nih.gov/compound/metformin (accessed on 12 May 2021)], the pKa of metformin is 12.4. This value indicates that metformin exists in a monoprotonated form at neutral pH (6–8), and that only when it reaches a pH < 3.1 would it exist as biprotonated species. Due to its unusually hydrophilic nature, metformin cannot passively diffuse through cell membranes, and must rely on members of organic cation transporters (OCTs) [1,2,3]. Therefore, only those tissues that express members of the OCT family (e.g., liver, kidney, and small intestine) will be targets for the action of metformin [5,6,7]. Metformin can also act on neuronal cells as they express two members of the OCT family, namely OCT1 and plasma membrane monoamine transporter (PMAT) [8]. Once metformin enters cells, it accumulates within the mitochondria [1] (Figure 1).
The therapeutic potential of metformin treatment expands its prescribed use as an anti-diabetic drug. Thus, metformin has been shown to be effective in the treatment of multiple diseases, including polycystic ovary syndrome [9], cardiovascular disease [10,11,12], and cancer [12,13,14]. In addition, it delays the aging process [12,15,16] and alleviates inflammation associated with it [17], and also modulates the microbiota to promote health [3,18]. Furthermore, metformin may have additional beneficial effects yet to be discovered. The beneficial effects of metformin also extend to neurodegenerative diseases; metformin has been reported to alleviate the pathophysiology of Alzheimer’s, Parkinson’s, and Huntington’s diseases, as well as multiple sclerosis, among others [12,19]. In this work, we will review the different proposed mechanisms of action of metformin and the beneficial effect that this drug has as a neuroprotective agent in different neurological diseases, with special attention to epileptic disorders and Lafora disease, a form of progressive myoclonus epilepsy (see below).
2. Proposed Mechanism of Action of Metformin to Lower Glucose Levels
2.1. Inhibition of Mitochondrial Glycerol-3-Phosphate Dehydrogenase (GPD2)
It is now widely accepted that the anti-hyperglycemic effect of metformin is mainly due to the suppression of hepatic glucose production (HGP) (reviewed in [1,2,3]). Hepatic glucose production is the result of the balance between glucose forming pathways (gluconeogenesis and glycogenolysis) and glucose consuming pathways (glycogen synthesis, glycolysis, and the pentose phosphate pathway) (Figure 1). Among them, hepatic gluconeogenesis contributes more than 50% of HGP and is considered the main pathway regulated by metformin. Metformin negatively regulates gluconeogenesis at different levels:
- (a)
- At the transcriptional level: metformin prevents cAMP responsive binding (CREB)-mediated transcription of the gluconeogenic glucose 6-phosphatase (G6PC) and phosphoenolpyruvate carboxykinase 1 (PEPCK1) genes. This is an indirect effect due to the inhibition of mitochondrial complex I by metformin (see below), leading to an increase in AMP levels, which inhibits adenylate cyclase and thus leads to a decrease in levels of cAMP, a mediator of CREB-dependent transcription. In addition, metformin has been proposed to activate AMP-activated protein kinase (AMPK), which has a negative effect on the transcriptional regulation of gluconeogenesis genes, among others [4] (see below);
- (b)
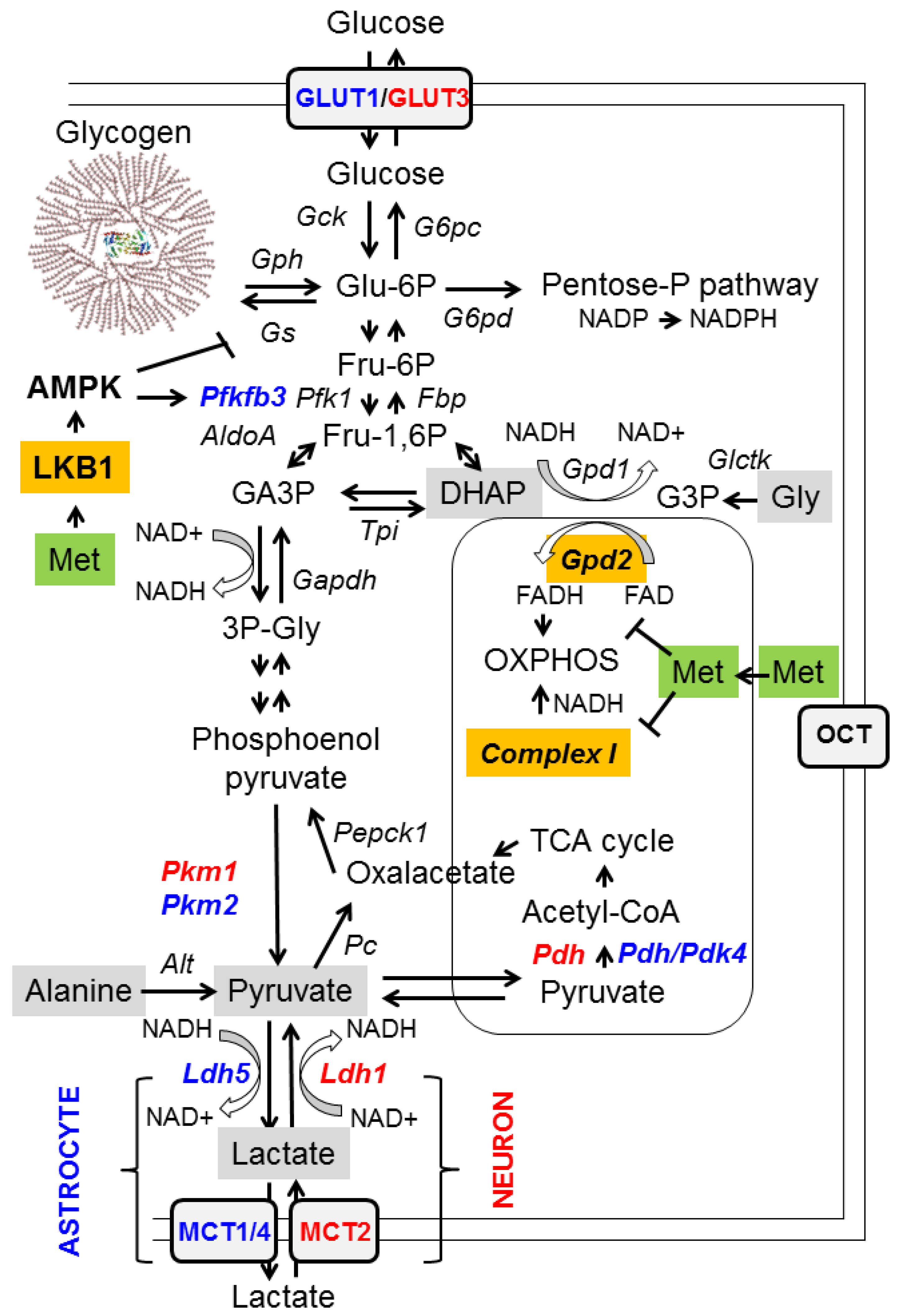
- Reducing the availability of gluconeogenic substrates: hepatic gluconeogenesis depends on the availability of appropriate substrates, such as glycerol, lactate, pyruvate, alanine, and dihydroxyacetone phosphate (DHAP), in order to convert them to glucose (Figure 1, grey boxes). Glycerol and DHAP are mutually interconnected, since glycerol is converted to glycerol 3-P (G3P) by glycerate kinase (Glctk) and then G3P is converted to DHAP by mitochondrial glycerol-3-phosphate dehydrogenase (GPD2). Metformin has been shown to inhibit mitochondrial GPD2 at regular concentrations (50–100 μM) (reviewed in [1,2,3]; Figure 1, orange box). Therefore, after treatment with metformin, the levels of DHAP are reduced, and this leads to a decrease in the flux of gluconeogenesis. As a consequence of this inhibition, G3P and glycerol accumulate in hepatocytes (Figure 1).
The function of GPD2 is coupled to one of the major NADH/NAD+ shuttles, the alpha-glycerophosphate shuttle, which consumes NADH and transforms DHAP to glycerol-3P through the action of cytosolic glycerol-3P dehydrogenase 1 (GPD1). Inhibition of GPD2 by metformin alters the cytosolic redox balance and leads to a higher NADH/NAD+ ratio, due to low levels of DHAP. This high NADH/NAD+ ratio also prevents the conversion of lactate into pyruvate by lactate dehydrogenase (LDH). Therefore, inhibition of GPD2 by metformin decreases the levels of two of the main gluconeogenic substrates, DHAP and pyruvate, and leads to the accumulation of glycerol and lactate (Figure 1). This accumulation of lactate is probably the cause of the appearance of lactic acidosis in some patients treated with metformin [1,2,3].
However, metformin does not affect the gluconeogenic use of pyruvate and alanine as substrates, since their entry into the gluconeogenesis pathway does not involve a redox-dependent mechanism (Figure 1). This would explain why hypoglycemia is rarely observed in patients treated with metformin or in healthy individuals, since part of the gluconeogenesis pathway is still active [1].
2.2. Inhibition of Mitochondrial Complex I of the Respiratory Chain and Activation of AMP-Activated Protein Kinase (AMPK)
An additional mechanism to explain the hypoglycemic effect of metformin on hepatocytes is its inhibitory action on mitochondrial complex I (NADH/ubiquinone oxidoreductase) [20]. Mitochondrial complex I of the respiratory chain is the site of the contribution of NADH to the proton gradient of OXPHOS (oxidative phosphorylation) (Figure 1, orange box). Inhibition of complex I by metformin reduces the mitochondria’s ability to consume NADH and the production of ATP. High levels of NADH and low levels of ATP have a crucial negative impact on the gluconeogenesis pathway, as this process requires a large amount of energy and depends on a correct NADH/NAD + balance. Furthermore, as ATP production is reduced, the AMP/ATP ratio increases, and this leads to the activation of AMP-activated protein kinase (AMPK), a master regulator of energy homeostasis [2,4,21]. AMPK activation leads to the activation of catabolic pathways (e.g., glycolysis through the activation of Pfkfb3, an enzyme involved in the formation of 2,6-fructose bisphosphate, an allosteric activator of phosphofructokinase 1 (Pfk1)) and the inhibition of anabolic pathways (e.g., glycogen synthesis by inhibiting glycogen synthase; Gs), to restore energy balance (Figure 1) [4,21]. AMPK exerts this function both at the transcriptional level, regulating the activity of different transcriptional factors by phosphorylation (e.g., the downregulation of CREB, carbohydrate-responsive element binding protein (ChREBP), and sterol regulatory element binding protein (SREBP-1), which are involved in the expression of genes related to gluconeogenesis, the carbohydrate metabolism, and sterol biosynthesis, respectively; on the other hand, AMPK upregulates peroxisome proliferator-activated receptor γ co-activator 1 alpha (PGC1alpha), involved in mitochondrial biogenesis, and activates pro-health span molecules, such as the forkhead box O3 (FOXO3) transcription factor and sirtuin 1 (SIRT1) deacetylase, which in turn induce the expression of protective molecules [2,4,21,22]). AMPK also operates at the level of key metabolic enzyme activity (e.g., inhibition of acetyl-Co carboxylase (Acc1/2), an enzyme involved in the synthesis of malonyl-CoA, an intermediate in fatty acid synthesis, and an inhibitor of fatty acid oxidation; therefore, AMPK activation inhibits the synthesis of fatty acids and promotes their degradation) [21]. Therefore, the increase in the AMP/ATP ratio caused by metformin activates AMPK indirectly, but this effect is only obtained when metformin is administered at supra-pharmacological concentrations (>1 mM) [1]. Recent results support the indirect effect of metformin on the activation of AMPK, as they show that metformin does not affect AMPK directly, but acts on the upstream liver kinase (LKB1), which participates in the phosphorylation and activation of the catalytic alpha subunit of AMPK [23] (Figure 1, orange box).
2.3. Effects of Metformin on Glucose Metabolism in the Brain
The effect of metformin on glucose metabolism occurs not only in hepatocytes, but also in other tissues, such as the brain [4]. Neurons and astrocytes are recognized to have different glucose metabolic profiles. The metabolism of neurons relies more on mitochondrial oxidative phosphorylation (OXPHOS) [24,25], due to the singularities of different glycolytic enzymes present in these cells. Thus, neurons have a pyruvate kinase isoform (Pkm1) that is completely active, and the resulting pyruvate is rapidly transformed into acetyl-CoA by active mitochondrial pyruvate dehydrogenase (Pdh) (Figure 1, enzymes highlighted in red) [24,25]. On the other hand, neurons express a lactate dehydrogenase isoform (Ldh1) that works better by transforming lactate into pyruvate, so the amount of lactate produced by them is very low. Furthermore, they express the monocarboxylate transporter 2 (MCT2) which is fully active in the uptake of lactate from the surrounding medium [24,25] (Figure 1, enzymes highlighted in red). In conclusion, neurons are prepared to capture lactate and convert it into pyruvate, which will enter the mitochondria to obtain energy. In contrast, astrocytes have a glucose metabolism that is more glycolytic [24,25]. They have reduced OXPHOS due to the low activity of their mitochondrial pyruvate dehydrogenase (Pdh), since it is inactivated by an active pyruvate dehydrogenase kinase 4 (Pdk4) (Figure 1, enzymes highlighted in blue). In addition, astrocytes express an isoform of lactate dehydrogenase (Ldh5) that is fully active in reducing pyruvate to lactate, which is exported outside the cell by monocarboxylate transporters MCT1/4. Hence, the glucose metabolism of astrocytes is designed to produce lactate and export it to the surrounding media. In addition, astrocytes can enhance glycolytic flux, because they have an active phosphofructokinase 2,6-bisphosphatase (Pfkfb3) that synthesizes 2,6-fructose bisphosphate, a potent allosteric activator of phosphofructokinase 1 (Pfk1) (Figure 1, enzymes highlighted in blue), and this enzyme can be fully activated by AMPK [24,25] (see above).
Bearing all this information in mind, we hypothesize that at the central nervous system (CNS), metformin would produce a reduction in gluconeogenesis in both neurons and astrocytes by inhibiting mitochondrial Gpd2 and activating AMPK, which would cause a decrease in the levels of DHAP and pyruvate, as in the case of hepatocytes (see above); in addition, this would cause an increase in the glycolytic flux (use of Glu-6P). This would in turn lead to increased lactate production in astrocytes and increased OXPHOS in neurons. Glycolysis would also increase in astrocytes, due to the metformin-mediated activation of AMPK that would activate Pfkfb3, leading to an enhancement of Pfk1 activity (Figure 1). To cope with a higher demand for Glu-6P, glucose uptake and glycogen degradation would be accelerated. As metformin-mediated activation of AMPK would also inactivate glycogen synthase, we expect glycogen levels to decrease after metformin treatment. This could explain the prevention in polyglucosan synthesis in mouse models of Lafora disease after treatment with metformin [26] (see below).
2.4. Metformin Ameliorates Oxidative Stress
Substantial evidence shows that metformin exerts antioxidant effects. Some of these can be attributed to the inhibition of mitochondrial complex I, which reduces reactive oxygen species (ROS) production by the OXPHOS respiratory chain [4]. In addition, metformin has other functions related to the activation of the AMPK pathway: (i) reduction of ROS levels by upregulating the expression of antioxidant enzymes, such as thioredoxin, through the AMPK–FOXO3 pathway; (ii) modulation of the expression of sirtuin 3 (SIRT3) deacetylase, whose activity promotes antioxidant effects in the cell; (iii) downregulation of NADPH oxidase, one of the main producers of cellular ROS; and (iv) enhancement of mitochondrial biogenesis by enhancing the function of PGC1alpha transcription factor [27].
2.5. Metformin and Neuroinflammation
Following brain injury, neuroinflammation is initially neuroprotective, but when it becomes chronic or excessive, it eventually causes damage [28]. It is now accepted that sustained brain inflammation promotes neuronal hyperexcitability and seizures, and that dysregulation in the immunoinflammatory function of the glia is a common factor that predisposes or contributes to the generation of seizures. At the same time, acute seizures upregulate the production of pro-inflammatory cytokines in microglia and astrocytes, triggering a cascade of inflammatory mediators. Thus, epileptic seizures and inflammatory mediators form a positive feedback loop, reinforcing each other [28]. For this reason, it has recently been proposed that the treatment of inflammation with specific anti-inflammatory drugs may be beneficial in the treatment of refractory epilepsies [28]. However, since the use of general anti-inflammatory drugs is not recommended due to their detrimental performance in long-term treatments [29], only specific anti-inflammatory compounds, whose selection has been made after a thorough understanding of the main related inflammatory pathways, should be used with each particular type of epilepsy.
Activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) is a hallmark of neuroinflammation, and is present in most neurological diseases. The Toll-like receptor 4 (TLR4) signaling pathway induces NF-kB activation through myeloid differentiation primary response 88 (MyD88) and tumor necrosis receptor-associated factor 6 (TRAF6), leading to the expression of pro-inflammatory mediators: cytokines, chemokines, cyclooxygenase 2 (COX2), and inducible nitric oxide synthase (iNOS) [30]. It has been described that the activation of AMPK by metformin reduces general inflammatory conditions since it inhibits the signaling of NF-kB, as well as the expression of pro-inflammatory cytokines (interleukin 1-beta (IL-1beta), interleukin 6 (IL-6), tumor necrosis factor alpha (TNFalpha), C–C motif chemokine ligand 2 (CCL2), etc.) in different cell types [31,32,33], suggesting that AMPK activation could protect against neuroinflammation [19]. Similarly, activation of AMPK by berberine reduces activated microglia; neutrophil infiltration; and IL-1beta, IL-6, CCL2, and CXCL2 production, which occur after traumatic brain injury [34]. In both cases, AMPK prevented the activation of the TLR4/NF-kB signaling pathway [31,34,35]. AMPK also inhibits lipopolysaccharide (LPS)-induced expression of proinflammatory cytokines (TNF-alpha, IL-1beta, and IL-6) by attenuating LPS-induced, TLR4-mediated NF-kB activation [36,37,38]. Similarly, AMPK prevented the advanced glycation end-product (AGE)-mediated signaling pathway, which ends with an increase in NF-kB expression and reduced iNOS and COX2 levels in AGE-treated human neural stem cells (hNSCs) [39]. The anti-inflammatory action of AMPK was also associated with the inhibition of LPS-induced activation of the phosphatidyl inositol 3 kinase (PI3-kinase)/RAC-alpha serine/threonine-protein kinase (Akt) pathway [37]. Downregulation of NF-kB levels inhibits the activation of the nucleotide-binding oligomerization domain and leucine-rich repeat and pyrin domain 3 (NLRP3) inflammasomes, while decreasing the activation of caspase1 and reducing the production of IL-1beta [38]. These mechanisms are particularly important in microglia, where AMPK inhibits the release of pro-inflammatory markers, decreasing neuroinflammation [40], and in astrocytes, where AMPK inhibits elevated ER stress and hyperglycemia-induced inflammation [41].
Since neuroinflammation is a recognized event associated with neurological disorders, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, and epilepsy, metformin could have a positive effect on these diseases (see below).
3. Metformin as a Neuroprotective Agent in Different Neurological Disorders
There is growing interest in the potential use of metformin in diseases of the central nervous system (CNS). Although most neurological disorders are different in nature, they share basic pathological mechanisms that are altered in the corresponding disease. Examples of these are the AMPK and mechanistic target of rapamycin (mTOR) kinase pathways. As indicated above, AMPK is a master regulator of energy homeostasis. It is activated in conditions of energy deprivation, and by activating catabolic pathways and inhibiting anabolic pathways it restores energy balance. In contrast, the mTOR pathway is activated under high-energy conditions, and operates by activating anabolic pathways and inhibiting catabolic pathways. Both the AMPK and mTOR pathways are interconnected, and activation of the AMPK pathway results in inhibition of the mTOR system, either by directly inactivating components of the mTOR complex (e.g., raptor, tuberous sclerosis complex 2 (TSC2)) or by reversing the effect of mTOR on common substrates (e.g., ULK1 in the autophagy process) [42]. In particular, metformin activates AMPK signaling, and also inhibits mTOR signaling via AMPK-dependent as well as AMPK-independent pathways [1,2,3]. In the following, we will briefly review the action of metformin as a neuroprotector agent in various neurological disorders.
3.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder. It is characterized by progressive memory loss and impaired cognitive function. Clinical studies indicate an ameliorative effect of metformin on cognitive decline and Alzheimer’s disease [43]. In fact, patients with T2D receiving metformin had a lower risk of cognitive impairment and a lower risk of developing Alzheimer’s disease than other patients with T2D receiving alternative treatments [44]. AMPK activation has also been reported to have neuroprotective effects in different mouse models of Alzheimer’s disease [45,46].
3.2. Parkinson’s Disease
Parkinson’s disease is the second most prevalent neurodegenerative disorder. Disease progression has been considered a consequence of mitochondrial dysfunction, with elevated levels of reactive oxygen species (ROS) and increased oxidative stress leading to the death of dopaminergic neurons [47]. Treatment with metformin reduces oxidative stress and improves the expression of antioxidant enzymes, such as superoxide dismutase and catalase [48]. This effect of metformin was achieved both by activating the AMPK pathway as well as through AMPK-independent mechanisms, such as activation of the brain-derived neurotrophic factor (BDNF) signaling pathway [49]. In animal models of Parkinson’s disease, metformin inhibits alpha-synuclein aggregation, prevents mitochondrial dysfunction, attenuates oxidative stress, enhances autophagy through AMPK activation, and prevents neurodegeneration and neuroinflammation (reviewed in [50]).
3.3. Huntington’s Disease
Huntington’s disease is an autosomal, dominant inherited disorder related to the presence of a defective huntingtin gene (Htt) [51]. Mutant huntingtin protein containing long poly-Q tracks overloads the ubiquitin proteasomal degradation system and forms aggregates with itself and with other proteins, leading to a depletion of critical molecules involved in neuronal homeostasis and resulting in neuronal degeneration [51]. Treatment with metformin has been reported to reduce the number of huntingtin aggregates, probably due to activation of autophagy, which results from activation of the AMPK pathway [43].
The reduction in the number of huntingtin aggregates is correlated with an improvement in cognitive and behavioral function in mouse models of Huntington’s disease [52].
3.4. Multiple Sclerosis
Multiple sclerosis is a chronic autoimmune disease that causes demyelination and destruction of neuronal cells at the CNS [53]. Current therapeutic approaches are based on the regulation of autoimmune attacks and the preservation of oligodendrocyte function. Metformin alleviates oxidative stress and restores mitochondrial function in patients with multiple sclerosis [54,55]. Furthermore, in an AMPK-dependent manner, metformin was able to enhance the expression of genes involved in the protection of oligodendrocytes and the restoration of central nervous system functions in an experimental model of autoimmune encephalomyelitis [56].
3.5. Epilepsy
Epilepsy is a neurological disorder characterized by a predisposition to generate epileptic seizures and the associated cognitive, psychological, and social consequences of this condition [28,57]. Epilepsy affects 1% of the total world population (around 65 million people worldwide) and is caused by acquired injuries in the brain (for example, after a stroke or traumatic brain injury), infectious diseases, autoimmune diseases, and genetic mutations. To date, more than 500 genes are associated with epilepsy [57]. The first-line treatment for epilepsy is antiseizure drugs (ASDs). The development of ASDs was based on the neuron-centric hypothesis that an imbalance of excitatory and inhibitory currents was largely responsible for epileptic seizures [58]. However, despite the availability of many ASDs, approximately one-third of patients fail to control seizures or soon become resistant to the effects of the ASDs [57,58]. Consequently, there is a critical need to develop innovative antiepileptic treatment strategies to improve progression and limit the detrimental consequences of the disease.
Since some forms of epilepsy are related to the upregulation of the mTOR pathway, different therapeutic strategies have been designed to inhibit this pathway [59]. The inhibition of mTOR signaling promotes a reduction in the generation of pro-inflammatory cytokines and chemokines (IL-1beta, TNFalpha, CCL2, iNOS, etc.) by microglia, which results in improvements in the motor deficit in the middle cerebral artery occlusion (MCAO) model of cerebral stroke [60]. As indicated above, AMPK is a master regulator of energy homeostasis: it is activated under conditions of energy deprivation, and by activating catabolic and inhibiting anabolic pathways it restores energy balance. In contrast, the mTOR pathway is activated under high-energy conditions, and operates by activating anabolic pathways and inhibiting catabolic pathways. Activation of these pathways occurs at the CNS, as with other peripheral tissues [4]. Interestingly, the AMPK and mTOR pathways are interconnected, and the activation of the AMPK pathway results in the inhibition of the mTOR system (see above). In particular, activation of AMPK by metformin inhibits mTOR signaling and has resulted in improved seizure control in models of mTOR overactivation [1,3,61,62].
These results were confirmed by a recent report indicating that AMPK activation by metformin improved lithium- and pilocarpine-induced status epilepticus in rats by inhibiting the mTOR pathway [63]. The beneficial effect of AMPK activation by metformin and calorie restriction on the main symptoms of an electric-ignition model of epilepsy, especially those related to generalized seizures, has also been observed [64].
Using an epileptic diabetic rat model, the activation of AMPK by metformin was reported to improve the inflammatory status and histopathological alterations present in this model [65]. Metformin normalized the levels of glutamate and gamma-aminobutyric acid (GABA), and reduced the levels of IL-1beta, TNF-alpha, NF-kB, and caspase 3. These effects were proposed to be a consequence of AMPK activation and mTOR inhibition [65].
AMPK activation also has beneficial effects on other models of epilepsy. For example, metformin has a beneficial effect against seizures and epilepsy in mouse models of induced epilepsy with pentylenetetrazol (PTZ), a pro-convulsive agent, due to its antioxidant and anti-apoptotic actions and upregulation of heat shock protein 70 (Hsp70) [62,66,67]. The anti-apoptotic effect was related to a downregulation of the RNA-like endoplasmic reticulum kinase (PERK) pro-apoptotic protein kinase/eukaryotic initiation factor 2 alpha (eIF2alpha)/activating transcription factor 4 (ATF4)/C/EBP homologous protein (CHOP) pathway [39]. Recently, it has been described that metformin reduces CHOP expression and apoptosis induced by status epilepticus in rats [68]. The beneficial effects of metformin on this model could also be due to an improvement in autophagy [69]. Additional reports indicate that AMPK activation by metformin decreases seizure susceptibility, facilitates seizure termination, and reduces the number and duration of seizures in a PTZ-induced epilepsy model [70].
AMPK activation by metformin also exhibits an anti-inflammatory effect in kainate-induced status epilepticus by inhibiting IL-1beta production and reducing the expression of glial fibrillary acidic protein (GFAP) and S100beta markers of astrogliosis, as well as by increasing secretion of the anti-inflammatory cytokine IL-10 [71]. In this model, metformin exerted a neuroprotective role against the kainate-induced epileptogenic process by preventing neuronal cell death, aberrant neurogenesis, and mossy fiber sprouting [72].
An additional relationship between AMPK and epilepsy is seen through the regulatory action on glucose transporter GLUT1. GLUT1 is the main glucose transporter in endothelial and astrocytic cells. GLUT1 deficiencies disrupt glucose transport in the brain, and this leads to seizures in patients with GLUT1 deficiency syndrome (OMIM 606777) [73]. AMPK activation has recently been shown to regulate the translocation of GLUT1 to the plasma membrane from internal stores by destabilizing the thioredoxin-interacting protein (TXNIP). This enhances astrocytic glucose uptake and glycolysis and enables proper regulation of the astrocyte–neuron lactate shuttle (ANLS), which preserves neuronal metabolic functionality [74]. This could explain the beneficial effects of AMPK activation by metformin on PTZ-induced seizures in mice on a high-fat diet, since after treatment with metformin, a normalization in the expression levels of GLUT1 and GLUT3 was observed [75].
In conclusion, AMPK activation was able to attenuate the generation of seizures by delaying the onset of epilepsy, reducing neuronal loss in the hippocampus, and preventing cognitive impairments in both acute and chronic epilepsy models. In the case of metformin, its antiepileptic effects could be attributed to an amelioration of oxidative brain damage, activation of the AMPK pathway, inhibition of the mTOR signaling, downregulation of brain-derived neurotrophic factor (BDNF), and neurotrophic receptor tyrosine kinase 2 (TrkB) levels or improvement of proteostasis [76,77].
Therefore, AMPK can be considered as a new anti-inflammatory and antiepileptic signaling pathway. Its activation leads to a decrease in the mTOR and TLR4/NF-kB signaling pathways, thus representing a promising therapeutic target for immunoinflammatory disorders like epilepsy [78]. In fact, in a recent screening of repurposing drugs with anticonvulsive properties, AMPK activators, such as metformin, appeared as promising candidates with therapeutic potential as anti-epileptic drugs [79].
4. Metformin and Lafora Disease
Lafora disease (OMIM 254780) is an ultra-rare form of autosomal, recessive progressive myoclonic epilepsy (PME), characterized by the accumulation of insoluble glycogen-like inclusions (polyglucosans) in the brain and other tissues; these are called Lafora bodies (LBs), and they can be identified with periodic acid Schiff (PAS) staining [80,81,82,83,84,85,86]. Lafora is a neurodegenerative disease, also considered a glycogen storage disease [87,88,89,90,91,92]. Various genetically engineered animal models, showing multiple symptoms present in patients with the disease, have been used to study the molecular basis of the disorder and search for effective therapies [93,94,95,96,97]. One of the compounds that successfully improved their symptoms is metformin [26,98], and as we will see below, it is already being used in clinical practice.
4.1. Clinical Aspects of Lafora Disease
Lafora disease is a fatal neurological disorder that usually begins in children between the ages of 10 and 15 who appear to have normal neurological development. The first symptoms are epileptic seizures, myoclonus, and/or cognitive alterations that cause school difficulties [86,99]. The cognitive and neurological deterioration is very rapid, and very soon language and intellectual problems occur that continue to worsen until patients develop severe dementia, ataxia, dysarthria, amaurosis, and respiratory failure. There are no specific treatments for the disease; seizures alone are treated with anticonvulsant drugs, but patients quickly develop resistance and myoclonus becomes constant. Status epilepticus or aspiration pneumonitis usually leads to death within the next 5 to 10 years from diagnosis [86,100,101].
Mutations in EPM2A (epilepsy of progressive myoclonus type 2 gene A) or NHLRC1/EPM2B (NHL repeat-containing protein 1/epilepsy of progressive myoclonus type 2 gene B) [102,103,104,105], encoding the laforin or malin proteins, respectively [106,107,108], have been described as causes of the disease. Lafora disease-causing mutations in the EPM2A gene represent approximately 60% and in the EPM2B gene 35% of patients with Lafora disease. Patients with mutations in the EPM2B gene appear to have a slightly milder phenotype than patients with mutations in the EPM2A gene [109]. In addition, mutations in the same gene and even the same mutation have been described to be associated with important phenotypic variations. Therefore, it has been suggested that genetic or epigenetic modifying factors could be responsible for the age of onset and severity of the disease [110].
Laforin and malin form a functional complex and work together in the regulation of glycogen synthesis, in the homeostasis of glucose transporters, in the maintenance of proteostasis, and in the response to oxidative stress, among other physiological pathways (reviewed in [111]).
4.2. Animal Models of Lafora Disease
Several experimental models of Lafora disease have been generated by deleting the Epm2a or Epm2b genes in mice. We and other groups have analyzed the functional alterations of two of them: Epm2a−/− [93] and Epm2b−/− [97] mice. Both models present memory deficiencies, impaired motor activity and coordination, dyskinesia, altered neuronal excitability, and myoclonus similar to those present in patients with the disease [93,97,112,113]. The presence of neurodegenerative processes and the accumulation of Lafora bodies in the brain correlate with the appearance of functional abnormalities and with the presence of reactive astrogliosis, oxidative stress, altered proteostasis, and impaired autophagy [30,97,114,115,116,117,118,119,120,121].
4.3. Pharmacological Interventions in Animal Models of Lafora Disease
We have studied the effect of various pharmacological treatments in the Epm2b−/− mouse model [26,98,122]. To improve proteostasis, we use 4-phenylbutyric acid (4-PBA), a chemical chaperone that reverses the misfolding and aggregation of proteins associated with various neurodegenerative diseases [123,124]; trehalose, another chemical chaperone that prevents protein denaturation and protects cellular integrity against stress phenomena [125]; and sodium selenate, an agent that reduces oxidative stress and the appearance of epileptic seizures in other animal models [126,127]. Our results indicate that, of the substances used, 4-PBA and sodium selenate, considerably improve memory impairment, motor activity, abnormal posture, and dyskinesia, as well as sensitivity to PTZ [26,98,122].
We also use metformin in Epm2b−/− mice, and showed that it reduces the accumulation of polyglucosans and polyubiquitin aggregates in the brain, decreases neuronal loss and reactive astrogliosis, and improves neuropsychological tests [26]. In addition, metformin decreases susceptibility to seizures, reduces the number and length of seizures, and eliminates the mortality induced by the pro-convulsive agent pentylenetetrazol (PTZ) in Epm2a−/− and Epm2b−/− mice [98]. These results allowed the designation of metformin as an orphan drug for the treatment of Lafora disease by the European Medicines Agency (EMA) in 2016 and the United States Food and Drug Administration (FDA) in 2017.
Due to these regulatory authorizations, metformin has been introduced into the clinical treatment of patients with Lafora disease. In a study with 10 patients, the authors indicated that the result of the metformin administration was inconclusive, probably because the patients engaged in the trial were quite advanced in the disease. In any case, the authors suggested that treatments should be attempted as early as possible in the course of Lafora disease [128].
5. Conclusions
In conclusion, metformin has beneficial effects on several neurological disorders that could be attributed to both AMPK-dependent and AMPK-independent mechanisms of action (Figure 2).
As a whole, metformin improves mitochondrial function (thereby reducing ROS production and oxidative stress), reduces the inflammatory response, reduces glucose production, and improves proteostasis (enhances the degradation of toxic aggregates). Therefore, the use of metformin as a disease-modifying drug is widely recognized. However, as the regular dose of metformin is 1–2 g/day, future studies should be aimed to understand what is the main mechanism of action related to neuroprotection, in order to look for more active compounds that affect only this pathway.
Author Contributions
P.S. and M.P.S. wrote the paper; P.S. made the figures; J.M.S. reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Spanish Ministry of Science and Innovation –SAF2017-83151-R to P.S. and Rti2018-095784b-100 SAF to M.P.S. and J.M.S.—and a grant from the National Institutes of Health (P01 NS097197), which established the Lafora Epilepsy Cure Initiative (LECI), to P.S., J.M.S. and M.P.S.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- LaMoia, T.E.; Shulman, G.I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Demare, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing ampk to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Alnouti, Y.; Petrick, J.S.; Klaassen, C.D. Tissue distribution and ontogeny of organic cation transporters in mice. Drug Metab. Dispos. 2006, 34, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and autoimmunity: A “new deal” of an old drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, E.M.; Mendoza, S.; Hamer, T.; Sosa, F.; Glueck, C.J. Metformin therapy in polycystic ovary syndrome reduces hyperinsulinemia, insulin resistance, hyperandrogenemia, and systolic blood pressure, while facilitating normal menses and pregnancy. Metabolism 1994, 43, 647–654. [Google Scholar] [CrossRef]
- Lexis, C.P.; van der Horst-Schrivers, A.N.; Lipsic, E.; Valente, M.A.; Muller Kobold, A.C.; de Boer, R.A.; van Veldhuisen, D.J.; van der Harst, P.; van der Horst, I.C. The effect of metformin on cardiovascular risk profile in patients without diabetes presenting with acute myocardial infarction: Data from the glycometabolic intervention as adjunct to primary coronary intervention in st elevation myocardial infarction (gips-iii) trial. BMJ Open Diabetes Res. Care 2015, 3, e000090. [Google Scholar]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (removal): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; He, S.J.; Feng, X.; Cheng, J.; Luo, Y.T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Devel. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Li, D. Metformin as an antitumor agent in cancer prevention and treatment. J. Diabetes 2011, 3, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskovatska, V.; Storey, K.B.; Vaiserman, A.M.; Lushchak, O. The use of metformin to increase the human healthspan. Adv. Exp. Med. Biol. 2020, 1260, 319–332. [Google Scholar]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia 2017, 60, 1662–1667. [Google Scholar] [CrossRef] [Green Version]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex i. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Carling, D. Amp-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An ampk-foxo pathway mediates longevity induced by a novel method of dietary restriction in c. Elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. 2), 115–125. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of lafora disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Iannantuoni, F.; Gruevska, A.; Muntane, J.; Rocha, M.; Victor, V.M. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020, 34, 101517. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive glia inflammatory signaling pathways and epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ye, S.; Wang, S.; Sun, W.; Hu, Y. Metformin inhibits nuclear factor-kappab activation and inflammatory cytokines expression induced by high glucose via adenosine monophosphate-activated protein kinase activation in rat glomerular mesangial cells in vitro. Chin. Med. J. 2014, 127, 1755–1760. [Google Scholar] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowicz-Piasecka, M.; Sikora, J.; Szydlowska, A.; Skupien, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A future therapy for neurodegenerative diseases: Theme: Drug discovery, development and delivery in alzheimer’s disease guest editor: Davide brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hung, T.H.; Lee, C.Y.; Wang, L.F.; Wu, C.H.; Ke, C.H.; Chen, S.F. Berberine protects against neuronal damage via suppression of glia-mediated inflammation in traumatic brain injury. PLoS ONE 2014, 9, e115694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Tang, H.; Li, W.; Gong, Y.; Li, S.; Huang, J.; Fang, Y.; Yuan, W.; Liu, Y.; Wang, S.; et al. Ampk and its activator berberine in the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2020, 26, 5054–5066. [Google Scholar] [CrossRef]
- Giri, S.; Nath, N.; Smith, B.; Viollet, B.; Singh, A.K.; Singh, I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: A possible role of amp-activated protein kinase. J. Neurosci. 2004, 24, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, B.S.; Jin, Q.; Oh, Y.T.; Kim, S.S.; Kong, Y.; Cho, Y.H.; Ha, J.; Baik, H.H.; Kang, I. 5-aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced tnf-alpha production through inhibition of phosphatidylinositol 3-kinase/akt activation in raw 264.7 murine macrophages. Biochem. Biophys. Res. Commun. 2004, 318, 372–380. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. Amp-activated protein kinase inhibits nf-kappab signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.M.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Chen, Y.L.; Pei, D.; Lin, C.H.; Shih, Y.N.; Yen, C.H.; Chen, S.J.; et al. Metformin activation of ampk suppresses age-induced inflammatory response in hnscs. Exp. Cell Res. 2017, 352, 75–83. [Google Scholar] [CrossRef]
- Bayliss, J.A.; Lemus, M.B.; Santos, V.V.; Deo, M.; Davies, J.S.; Kemp, B.E.; Elsworth, J.D.; Andrews, Z.B. Metformin prevents nigrostriatal dopamine degeneration independent of ampk activation in dopamine neurons. PLoS ONE 2016, 11, e0159381. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin alleviates high glucose-induced er stress and inflammation by inhibiting the interaction between caveolin1 and ampkalpha in rat astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, J.L.; Hellberg, K.; Luo, E.C.; Van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. Ampk regulation of raptor and tsc2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. 2020, 34, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds that extend longevity are protective in neurodegenerative diseases and provide a novel treatment strategy for these devastating disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef] [PubMed]
- Orkaby, A.R.; Cho, K.; Cormack, J.; Gagnon, D.R.; Driver, J.A. Metformin vs sulfonylurea use and risk of dementia in us veterans aged >/= 65 years with diabetes. Neurology 2017, 89, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. Sirt1 deacetylase protects against neurodegeneration in models for alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in mptp-induced parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the mptp mouse model of parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging neuroprotective effect of metformin in parkinson’s disease: A molecular crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Sanchis, A.; Garcia-Gimeno, M.A.; Canada-Martinez, A.J.; Sequedo, M.D.; Millan, J.M.; Sanz, P.; Vazquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zq175 mouse model of huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.P.; Lin, Y.H.; Cui, Q.L.; Pernin, F.; Kennedy, T.E.; Ludwin, S.K.; Healy, L.M. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J. Neuroimmunol. 2019, 331, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, L.P.; Krotenko, N.V.; Grishko, E.V.; Krotenko, N.M.; Alifirova, V.M.; Ivanova, S.A. State of antioxidant system in patients with multiple sclerosis during therapy. Biomed. Khim. 2011, 57, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Bijak, M. Metformin as a potential agent in the treatment of multiple sclerosis. Int. J. Mol. Sci. 2020, 21, 5957. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Mohan, S.; Singh, A.K.; Singh, I. Amp-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am. J. Pathol. 2013, 183, 526–541. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Constanti, A.; Russo, E.; De Sarro, G. Mtor pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol. Res. 2016, 107, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.D.; Hao, J. Mtorc1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from m1 type to m2 type. FASEB J. 2016, 30, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of ampk, inhibits mtorc1 in a rag gtpase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Mehrabi, S.; Sanadgol, N.; Barati, M.; Shahbazi, A.; Vahabzadeh, G.; Barzroudi, M.; Seifi, M.; Gholipourmalekabadi, M.; Golab, F. Evaluation of metformin effects in the chronic phase of spontaneous seizures in pilocarpine model of temporal lobe epilepsy. Metab. Brain. Dis. 2018, 33, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bojja, S.L.; Medhi, B.; Anand, S.; Bhatia, A.; Joshi, R.; Minz, R.W. Metformin ameliorates the status epilepticus- induced hippocampal pathology through possible mtor modulation. Inflammopharmacology 2021, 29, 137–151. [Google Scholar] [CrossRef]
- Rubio Osornio, M.D.C.; Custodio Ramirez, V.; Calderon Gamez, D.; Paz Tres, C.; Carvajal Aguilera, K.G.; Phillips Farfan, B.V. Metformin plus caloric restriction show anti-epileptic effects mediated by mtor pathway inhibition. Cell Mol. Neurobiol. 2018, 38, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.A.E.; Abdel-Rahman, R.F.; Mahmoud, S.S.; Khattab, M.M.; Safar, M.M. Metformin and trimetazidine ameliorate diabetes-induced cognitive impediment in status epileptic rats. Epilepsy Behav. 2020, 104, 106893. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.R.; Xu, X.C.; Xu, F.; Zhang, W.L.; Liu, L.M.; Wang, W.P. Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole-induced kindling in mice. Biochem. Biophys. Res. Commun. 2014, 448, 414–417. [Google Scholar] [CrossRef]
- Hussein, A.M.; Eldosoky, M.; El-Shafey, M.; El-Mesery, M.; Ali, A.N.; Abbas, K.M.; Abulseoud, O.A. Effects of metformin on apoptosis and alpha-synuclein in a rat model of pentylenetetrazole-induced epilepsy. Can. J. Physiol. Pharmacol. 2019, 97, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, G.; Guo, H.; Shi, Z.N.; Jiang, J.; Wang, X.Y.; Yang, X.; Liu, X.Y. The effect of metformin treatment on endoplasmic reticulum (er) stress induced by status epilepticus (se) via the perk-eif2alpha-chop pathway. Bosn. J. Basic Med. Sci. 2018, 18, 49–54. [Google Scholar] [PubMed] [Green Version]
- Zeyghami, M.A.; Hesam, E.; Khadivar, P.; Hesam, H.K.; Ahmadnia, A.; Amini, A. Effects of atorvastatin and metformin on development of pentylenetetrazole-induced seizure in mice. Heliyon 2020, 6, e03761. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, B.; Zheng, F.; Li, Y.; Zhang, Y.; Hu, Y.; Wang, X. Chronic metformin treatment facilitates seizure termination. Biochem. Biophys. Res. Commun. 2017, 484, 450–455. [Google Scholar] [CrossRef]
- Vazifehkhah, S.; Khanizadeh, A.M.; Mojarad, T.B.; Nikbakht, F. The possible role of progranulin on anti-inflammatory effects of metformin in temporal lobe epilepsy. J. Chem. Neuroanat. 2020, 109, 101849. [Google Scholar] [CrossRef] [PubMed]
- Vazifehkhah, S.; Ali, M.K.; Babae, J.F.; Hashemi, P.; Alireza, M.S.; Nikbakht, F. Evaluation of the ameliorative effects of oral administration of metformin on epileptogenesis in the temporal lobe epilepsy model in rats. Life Sci. 2020, 257, 118066. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, R.; Gawali, M.V.; Tiwari, D.; Sukumaran, A.; Oatman, N.; Anderson, J.; Nardini, D.; Bhuiyan, M.A.N.; Tkac, I.; Ward, A.L.; et al. Ampk-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep. 2020, 32, 108092. [Google Scholar] [CrossRef] [PubMed]
- Nesci, V.; Russo, E.; Arcidiacono, B.; Citraro, R.; Tallarico, M.; Constanti, A.; Brunetti, A.; De Sarro, G.; Leo, A. Metabolic alterations predispose to seizure development in high-fat diet-treated mice: The role of metformin. Mol. Neurobiol. 2020, 57, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Nandini, H.S.; Paudel, Y.N.; Krishna, K.L. Envisioning the neuroprotective effect of metformin in experimental epilepsy: A portrait of molecular crosstalk. Life Sci. 2019, 233, 116686. [Google Scholar]
- Yimer, E.M.; Surur, A.; Wondafrash, D.Z.; Gebre, A.K. The effect of metformin in experimentally induced animal models of epileptic seizure. Behav. Neurol. 2019, 2019, 6234758. [Google Scholar] [CrossRef]
- Pilon, G.; Dallaire, P.; Marette, A. Inhibition of inducible nitric-oxide synthase by activators of amp-activated protein kinase: A new mechanism of action of insulin-sensitizing drugs. J. Biol. Chem. 2004, 279, 20767–20774. [Google Scholar] [CrossRef] [Green Version]
- Brueggeman, L.; Sturgeon, M.L.; Martin, R.M.; Grossbach, A.J.; Nagahama, Y.; Zhang, A.; Howard, M.A., 3rd; Kawasaki, H.; Wu, S.; Cornell, R.A.; et al. Drug repositioning in epilepsy reveals novel antiseizure candidates. Ann. Clin. Transl. Neurol. 2019, 6, 295–309. [Google Scholar] [CrossRef]
- Lafora, G.R.; Glueck, B. Beitrag zur histogpathologie der myoklonischen epilepsie. Gesamte Neurol. Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Harriman, D.G.; Millar, J.H.; Stevenson, A.C. Progressive familial myoclonic epilepsy in three families: Its clinical features and pathological basis. Brain 1955, 78, 325–349. [Google Scholar] [CrossRef]
- Schwarz, G.A.; Yanoff, M. Lafora’s disease. Distinct clinico-pathologic form of unverricht’s syndrome. Arch. Neurol. 1965, 12, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Austin, J.; Witmer, F.; Sakai, M. Studies in myoclonus epilepsy (lafora body form). I. Isolation and preliminary characterization of lafora bodies in two cases. Arch. Neurol. 1968, 19, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (lafora body form). Ii. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 1970, 20, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Karpati, G. Ultrastructural findings in lafora disease. Ann. Neurol. 1981, 10, 63–64. [Google Scholar] [CrossRef]
- Berkovic, S.F.; Andermann, F.; Carpenter, S.; Wolfe, L.S. Progressive myoclonus epilepsies: Specific causes and diagnosis. N. Engl. J. Med. 1986, 315, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.B. Corpora-amylacea and the family of polyglucosan diseases. Brain. Res. Brain. Res. Rev. 1999, 29, 265–295. [Google Scholar] [CrossRef]
- Raben, N.; Danon, M.; Lu, N.; Lee, E.; Shliselfeld, L.; Skurat, A.V.; Roach, P.J.; Lawrence, J.C., Jr.; Musumeci, O.; Shanske, S.; et al. Surprises of genetic engineering: A possible model of polyglucosan body disease. Neurology 2001, 56, 1739–1745. [Google Scholar] [CrossRef]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Roma-Mateo, C.; Sanz, P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein; et alii. FEBS J. 2013, 280, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Duran, J.; Guinovart, J.J. Brain glycogen in health and disease. Mol. Asp. Med. 2015, 46, 70–77. [Google Scholar] [CrossRef]
- Brewer, M.K.; Putaux, J.L.; Rondon, A.; Uittenbogaard, A.; Sullivan, M.A.; Gentry, M.S. Polyglucosan body structure in lafora disease. Carbohydr. Polym. 2020, 240, 116260. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the epm2a gene causes formation of lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaoli-Roach, A.A.; Tagliabracci, V.S.; Segvich, D.M.; Meyer, C.M.; Irimia, J.M.; Roach, P.J. Genetic depletion of the malin e3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 2010, 285, 25372–25381. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, J.; Wang, P.; Girard, J.M.; Ruggieri, A.; Wang, T.J.; Draginov, A.G.; Kameka, A.P.; Pencea, N.; Zhao, X.; Ackerley, C.A.; et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann. Neurol. 2010, 68, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Valles-Ortega, J.; Duran, J.; Garcia-Rocha, M.; Bosch, C.; Saez, I.; Pujadas, L.; Serafin, A.; Canas, X.; Soriano, E.; Delgado-Garcia, J.M.; et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of lafora disease. EMBO Mol. Med. 2011, 3, 667–681. [Google Scholar] [CrossRef]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanz, P.; Sanchez, M.P. 4-phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of lafora disease. Neuroreport 2017, 28, 268–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovic, S.F.; So, N.K.; Andermann, F. Progressive myoclonus epilepsies: Clinical and neurophysiological diagnosis. J. Clin. Neurophysiol. 1991, 8, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M. Idiopathic epilepsies with a complex mode of inheritance. Epilepsia 1999, 40 (Suppl. 3), 12–16. [Google Scholar] [CrossRef] [Green Version]
- Minassian, B.A. Lafora’s disease: Towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001, 25, 21–29. [Google Scholar] [CrossRef]
- Serratosa, J.M.; Delgado-Escueta, A.V.; Posada, I.; Shih, S.; Drury, I.; Berciano, J.; Zabala, J.A.; Antunez, M.C.; Sparkes, R.S. The gene for progressive myoclonus epilepsy of the lafora type maps to chromosome 6q. Hum. Mol. Genet. 1995, 4, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gomez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabe, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the lafora type (epm2). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in nhlrc1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Ganesh, S.; Agarwala, K.L.; Ueda, K.; Akagi, T.; Shoda, K.; Usui, T.; Hashikawa, T.; Osada, H.; Delgado-Escueta, A.V.; Yamakawa, K. Laforin, defective in the progressive myoclonus epilepsy of lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 2000, 9, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into lafora disease: Malin is an e3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006, 281, 30412–30418. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Abad, C.; Gómez-Garre, P.; Gutiérrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; Rodriguez de Cordoba, S.; Serratosa, J.M. Lafora disease due to epm2b mutations. A clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef]
- Jara-Prado, A.; Ochoa, A.; Alonso, M.E.; Lima Villeda, G.A.; Fernandez-Valverde, F.; Ruano-Calderon, L.; Vargas-Canas, S.; Duron, R.M.; Delgado-Escueta, A.V.; Martinez-Juarez, I.E. Late onset lafora disease and novel epm2a mutations: Breaking paradigms. Epilepsy Res. 2014, 108, 1501–1510. [Google Scholar] [CrossRef]
- Garcia-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora disease: A ubiquitination-related pathology. Cells 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cabrero, A.M.; Marinas, A.; Guerrero, R.; de Cordoba, S.R.; Serratosa, J.M.; Sanchez, M.P. Laforin and malin deletions in mice produce similar neurologic impairments. J. Neuropathol. Exp. Neurol. 2012, 71, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cabrero, A.M.; Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front. Neurosci. 2014, 8, 291. [Google Scholar] [PubMed] [Green Version]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Knecht, E.; Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado-Garcia, O.; Vernia, S.; Boya, P.; Sanz, P.; Rodriguez de Cordoba, S.; Rubinsztein, D.C. Impaired autophagy in lafora disease. Autophagy 2010, 6, 991–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, E.; Criado-Garcia, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; et al. Malin knockout mice support a primary role of autophagy in the pathogenesis of lafora disease. Autophagy 2012, 8, 701–703. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Roma-Mateo, C.; Aguado, C.; Garcia-Gimenez, J.L.; Knecht, E.; Sanz, P.; Pallardo, F.V. Oxidative stress, a new hallmark in the pathophysiology of lafora progressive myoclonus epilepsy. Free Radic. Biol. Med. 2015, 88, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Aguado, C.; Sanchez-Martin, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef] [Green Version]
- Espinos, C.; Galindo, M.I.; Garcia-Gimeno, M.A.; Ibanez-Cabellos, J.S.; Martinez-Rubio, D.; Millan, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; Garcia-Gimenez, J.L.; Moreno-Estelles, M.; Roma-Mateo, C.; Knecht, E.; Pallardo, F.V.; Sanz, P. Reactive glia-derived neuroinflammation: A novel hallmark in lafora progressive myoclonus epilepsy that progresses with age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of lafora disease. Epilepsia 2017, 58, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Frechilla, D.; Del Rio, J.; Garcia-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate up-regulates the dj-1 protein and protects neurons in cell culture and in animal models of parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, P.; Steinbrenner, H.; Sies, H. Selenium, oxidative stress, and health aspects. Mol. Asp. Med. 2005, 26, 256–267. [Google Scholar] [CrossRef]
- Rehni, A.K.; Singh, T.G. Selenium induced anticonvulsant effect: A potential role of prostaglandin e(1) receptor activation linked mechanism. J. Trace Elem. Med. Biol. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Bisulli, F.; Muccioli, L.; d’Orsi, G.; Canafoglia, L.; Freri, E.; Licchetta, L.; Mostacci, B.; Riguzzi, P.; Pondrelli, F.; Avolio, C.; et al. Treatment with metformin in twelve patients with lafora disease. Orphanet J. Rare Dis. 2019, 14, 149. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Metabolic pathways affected by metformin. A diagram of the main pathways related to glycolysis and gluconeogenesis is depicted. Gluconeogenic substrates are highlighted in grey, and enzymes directly affected by metformin are highlighted in orange. Specific enzyme isoforms present in neurons are in red, and those present in astrocytes are in blue. See text for details. AldoA: aldolase; Alt: Alanine aminotransferase; AMPK: AMP-activated protein kinase; NADH: ubiquinone oxidoreductase; Fbp: fructose bisphosphatase; Gapdh: glyceraldehyde-3P dehydrogenase; G6pc: glucose 6-phosphatase; G6pd: glucose-6P dehydrogenase; Gck: glucokinase; Glctk: glycerate kinase; GLUT: glucose transporter; Gpd: glycerol-3P dehydrogenase; Gph: glycogen phosphorylase; Gs: glycogen synthase; Ldh: lactate dehydrogenase; LKB1: liver protein kinase; MCT: monocarboxylic transporter; OCT: organic cation transporter; Pc: pyruvate carboxylase; Pdh: pyruvate dehydrogenase; Pdk: pyruvate dehydrogenase kinase; Pepck1: phosphoenolpyruvate carboxykinase; Pfk: phosphofructokinase; Pfkfb3: phosphofructokinase 2,6-bisphosphatase; Pkm: pyruvate kinase; TCA cycle: tricarboxylic acid cycle; Tpi: triosephosphate isomerase.
Figure 1.
Metabolic pathways affected by metformin. A diagram of the main pathways related to glycolysis and gluconeogenesis is depicted. Gluconeogenic substrates are highlighted in grey, and enzymes directly affected by metformin are highlighted in orange. Specific enzyme isoforms present in neurons are in red, and those present in astrocytes are in blue. See text for details. AldoA: aldolase; Alt: Alanine aminotransferase; AMPK: AMP-activated protein kinase; NADH: ubiquinone oxidoreductase; Fbp: fructose bisphosphatase; Gapdh: glyceraldehyde-3P dehydrogenase; G6pc: glucose 6-phosphatase; G6pd: glucose-6P dehydrogenase; Gck: glucokinase; Glctk: glycerate kinase; GLUT: glucose transporter; Gpd: glycerol-3P dehydrogenase; Gph: glycogen phosphorylase; Gs: glycogen synthase; Ldh: lactate dehydrogenase; LKB1: liver protein kinase; MCT: monocarboxylic transporter; OCT: organic cation transporter; Pc: pyruvate carboxylase; Pdh: pyruvate dehydrogenase; Pdk: pyruvate dehydrogenase kinase; Pepck1: phosphoenolpyruvate carboxykinase; Pfk: phosphofructokinase; Pfkfb3: phosphofructokinase 2,6-bisphosphatase; Pkm: pyruvate kinase; TCA cycle: tricarboxylic acid cycle; Tpi: triosephosphate isomerase.

Figure 2.
Mechanisms of action of metformin and cellular outcomes. Proteins affected by metformin, either via AMPK-dependent and AMPK-independent pathways, are indicated. In red are outcomes that are inhibited by metformin. In blue are those that are enhanced by metformin.
Figure 2.
Mechanisms of action of metformin and cellular outcomes. Proteins affected by metformin, either via AMPK-dependent and AMPK-independent pathways, are indicated. In red are outcomes that are inhibited by metformin. In blue are those that are enhanced by metformin.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sanz, P.; Serratosa, J.M.; Sánchez, M.P. Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. Int. J. Mol. Sci. 2021, 22, 5351. https://doi.org/10.3390/ijms22105351
AMA Style
Sanz P, Serratosa JM, Sánchez MP. Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. International Journal of Molecular Sciences. 2021; 22(10):5351. https://doi.org/10.3390/ijms22105351
Chicago/Turabian StyleSanz, Pascual, José Maria Serratosa, and Marina P. Sánchez. 2021. "Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease" International Journal of Molecular Sciences 22, no. 10: 5351. https://doi.org/10.3390/ijms22105351
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.