Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease


1
Laboratory of Mechanisms of Gene Expression, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Miklukho-Maklaya 16/10, 117997 Moscow, Russia
2
National Research Center “Kurchatov Institute”, 1, Academika Kurchatova pl., 123182 Moscow, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(21), 8055; https://doi.org/10.3390/ijms21218055
Submission received: 30 September 2020
/
Revised: 21 October 2020
/
Accepted: 22 October 2020
/
Published: 29 October 2020
(This article belongs to the Special Issue Medical Genetics, Genomics and Bioinformatics – 2020)
Abstract
:The neuronal ceroid lipofuscinoses (NCLs) collectively constitute one of the most common forms of inherited childhood-onset neurodegenerative disorders. They form a heterogeneous group of incurable lysosomal storage diseases that lead to blindness, motor deterioration, epilepsy, and dementia. Traditionally the NCL diseases were classified according to the age of disease onset (infantile, late-infantile, juvenile, and adult forms), with at least 13 different NCL varieties having been described at present. The current review focuses on classic juvenile NCL (JNCL) or the so-called Batten (Batten-Spielmeyer-Vogt; Spielmeyer-Sjogren) disease, which represents the most common and the most studied form of NCL, and is caused by mutations in the CLN3 gene located on human chromosome 16. Most JNCL patients carry the same 1.02-kb deletion in this gene, encoding an unusual transmembrane protein, CLN3, or battenin. Accordingly, the names CLN3-related neuronal ceroid lipofuscinosis or CLN3-disease sometimes have been used for this malady. Despite excessive in vitro and in vivo studies, the precise functions of the CLN3 protein and the JNCL disease mechanisms remain elusive and are the main subject of this review. Although the CLN3 gene is highly conserved in evolution of all mammalian species, detailed analysis of recent genomic and transcriptomic data indicates the presence of human-specific features of its expression, which are also under discussion. The main recorded to date changes in cell metabolism, to some extent contributing to the emergence and progression of JNCL disease, and human-specific molecular features of CLN3 gene expression are summarized and critically discussed with an emphasis on the possible molecular mechanisms of the malady appearance and progression.
1. Introduction
The juvenile neuronal ceroid lipofuscinosis (JNCL) or Batten disease is a devastating and fatal disorder that is connected to a mutation of the CLN3 (ceroid lipofuscinosis neuronal 3) gene. Up to 90% of patients with Batten disease carry the 1.02 kb deletion, which, in homozygous form, always causes a severe phenotype, including blindness, epilepsy, dementia, and early death at approximately 20–30 years of age [1]. There are a couple of excellent recent reviews on Batten disease, describing genetic basis [2] and molecular factors of the malady [3,4,5], scrupulously detailing the symptoms of the disease and its diagnosis [1] or summarizing data on what cellular processes are affected by CLN3 dysfunction [5], but a molecular explanation of how this well-characterized genetic defect (CLN3-mutations) affect neuronal physiology is still missing and the function(s) of the CLN3 protein is still elusive. As a rule (in more than 80% of patients), the disease begins with a distinct functional visual impairment and progressive vision loss at the age of 5–10 years, with complete blindness occurs at a later stage of the illness progression [6]. Fine granularity of the retinal pigment epithelium in the central macula and even bull’s-eye maculopathy may be present in the early stages of the disease.
When vision is affected, symptoms such as night blindness, tunnel vision (without peripheral vision) or vice versa—peripheral vision (without central vision), reticular vision, antipathy to bright light, slow adaptation from darkness to light and vice versa, blurred outlines of visible objects—i.e. blurred vision, poor color discrimination—come to the fore [7]. These features are explained by the relative preservation of the upper peripheral retina with the loss of the central and lower fields of vision and are usually manifested by a significantly reduced amplitude of the rod- and cone-response [8]. In the future, Batten disease develops atrophy of the optic nerve, weakening of blood vessels, and accumulation of pigment in the peripheral retina. Some JNCL patients were diagnosed with cataracts and even developed acute glaucoma in the later stages of the disease [9]. In general, the rate of progression of vision loss is extremely fast compared to retinal degeneration in other ophthalmic diseases and leads to legal blindness just a few years after presentation [7].
Progressive vision loss in previously healthy children is accompanied by personality changes, behavioral problems, and slow learning. Psychoneurological manifestations include a gradual loss of social adaptation with frequent and abrupt mood changes, calmness, clumsiness, stumbling, and lethargy [1]. These symptoms are replaced within 2–4 years by even more obvious psychomotor degeneration, leading to convulsive seizures and a persistent decrease in cognitive activity, followed by the loss of acquired skills (including walking, sitting, simple and necessary elements of self-service), loss of speech skills due to damage to the central speech motor apparatus (with understanding of the heard speech remains) [10,11]. This progressive loss of motor and learning (movement and speech) functions begins with Parkinson-like symptoms and leads to dementia. Eventually, those who suffer become disabled, bedridden, and die prematurely.
Batten disease is often difficult to diagnose because it is quite rare. The incidence of CLN3-disease in the United States and several European countries (for example, Germany) is approximately 1 per 100,000 live newborns [12]. The highest prevalence of JNCL is found in the Scandinavian countries, especially in Finland (slightly more than 1 in 25,000 live newborns with a carrier frequency of 1 in 70). Enrichment of certain alleles indicated that the same major mutation is responsible for Batten disease in Finland as in most other European countries. Moreover, haplotype analysis suggested that most CLN3-deficient chromosomes arose from a single founder mutation [13].
As noted above, vision problems are often the first symptoms. Therefore, the initial diagnosis can be made by an eye examination using the electrophysiological methods mentioned above. To confirm the diagnosis, blood and urine tests are performed to search for lipopigment buildup (e.g., searching for lymphocyte vacuoles in blood by light microscopy), as well as a skin biopsy with its examination under an electron microscope.
The next important step in confirming a diagnosis is to detect a mutation/deletion in the CLN3 gene. Munroe et al. used PCR to identify the intragenic microsatellite marker D16S298 to make the prenatal diagnosis of Batten disease on the basis of a chorionic villus sample [14]. A rapid diagnostic solid-phase minisequencing test to detect the most common 1.02-kb CLN3 deletion was developed in Finland [15]. Based on modern direct Sanger sequencing technique, more comprehensive diagnostic tests on all neuronal ceroid lipofuscinoses (NCLs) were recently developed [16]. Later manifested brain abnormalities are diagnosed using MRI scans, computed tomography and electroencephalogram (EEG) measurements.
Nearly half of JNCL patients have abnormalities in other organ and tissues. This includes, for example such cardio deficiencies as T-wave inversion, sinus node dysfunction and ventricular hypertrophy leading to severe bradycardia and/or other conduction abnormalities [17]. The skeletal muscles of all CLN3 patients show varying degrees of atrophy. They are homogeneously affected by an autophagic vacuolar myopathy (AVM) characterized by autophagic vacuoles and characteristic lysosomal pathology [18]. However, it is worth emphasizing that although the CLN3 gene is expressed everywhere, only certain tissues and organs of human body are affected, and neurological functions of the brain are the most severely damaged.
In this review, an attempt is made to examine all aspects of cellular physiology, which are disturbed in cells containing defective CLN3 protein and contribute to the development of pathological changes in the central nervous system, leading to the emergence and progression of still incurable Batten disease. The important human-specific aspect of expression and regulation of CLN3 gene is also considered.
2. Human-Specific Aspects of the CLN3 Gene Structure and Expression Which Could Have Influence on the Disease Progression
It was clearly established that juvenile neuronal ceroid lipofuscinosis (JNCL) or Batten disease (CLN3; OMIM #204200) is caused by homozygous or compound heterozygous mutation in the CLN3 gene (NCBI ID 1201, HGNC ID 2074, MIM ID 607042) located on the short arm of the Homo sapiens chromosome 16 at position 12.1 (16p12) [5]. In close vicinity to the CLN3 in the human genome, there are APOBR and IL27 genes encoding the apolipoprotein B receptor and interleukin 27 (on the right) and gene NPIPB7 of nuclear pore complex interacting protein family member B7 (even with partial overlap with CLN3) and still uncharacterized locus LOC112268171 (on the left).The mutation responsible for about 75% of JNCL disease is a genomic deletion of 1.02 kb, including 217 nt of the open reading frame (nucleotides 598–814) corresponding to two coding exons (number 7 and number 8) [19]. Elimination of these 217 bp of coding sequence produces a frameshift, generating a TAA termination codon 84 bp downstream of the deletion junction. The predicted translation product is a truncated protein of 181 amino acids consisting of the first 153 residues of the CLN3 protein, followed by 28 novel amino acids before the stop codon. Munroe et al. identified homozygosity for this common 1.02 kb CLN3 deletion in 139 (74%) of 188 unrelated patients with Batten disease [14]. The fact that most patients appear to be homozygous for a common CLN3 deletion fits with the prediction of CLN3 as not being haploinsufficient [20]. Mole and Cotman tabulated the mutations that had been identified in the various CLNs; they reported 67 mutations, 2 promoter changes and 13 polymorphisms associated with CLN3 [2]. The 181 amino acids long truncated protein produced from the common 1.02 kb deletion mutant allele retains significant CLN3 function, indicating that JNCL is a mutation-specific disease phenotype [21]. The residual function likely explains why classic Batten disease shows later onset and less severe clinical manifestations compared to other forms of neuronal ceroid lipofuscinoses (NLCs).
The full-length CLN3 protein, also called battenin, contains 438-amino acids. The amino acid sequence of the CLN3 gene product predicts a hydrophobic protein with 6 transmembrane domains [22]. The sequence appears to contain a putative mitochondrial targeting site at residue 11 and has four predicted N-glycosylation sites, all on the predicted exoplasmic surface. The CLN3 protein sequence does not display significant similarities to any protein of known function.
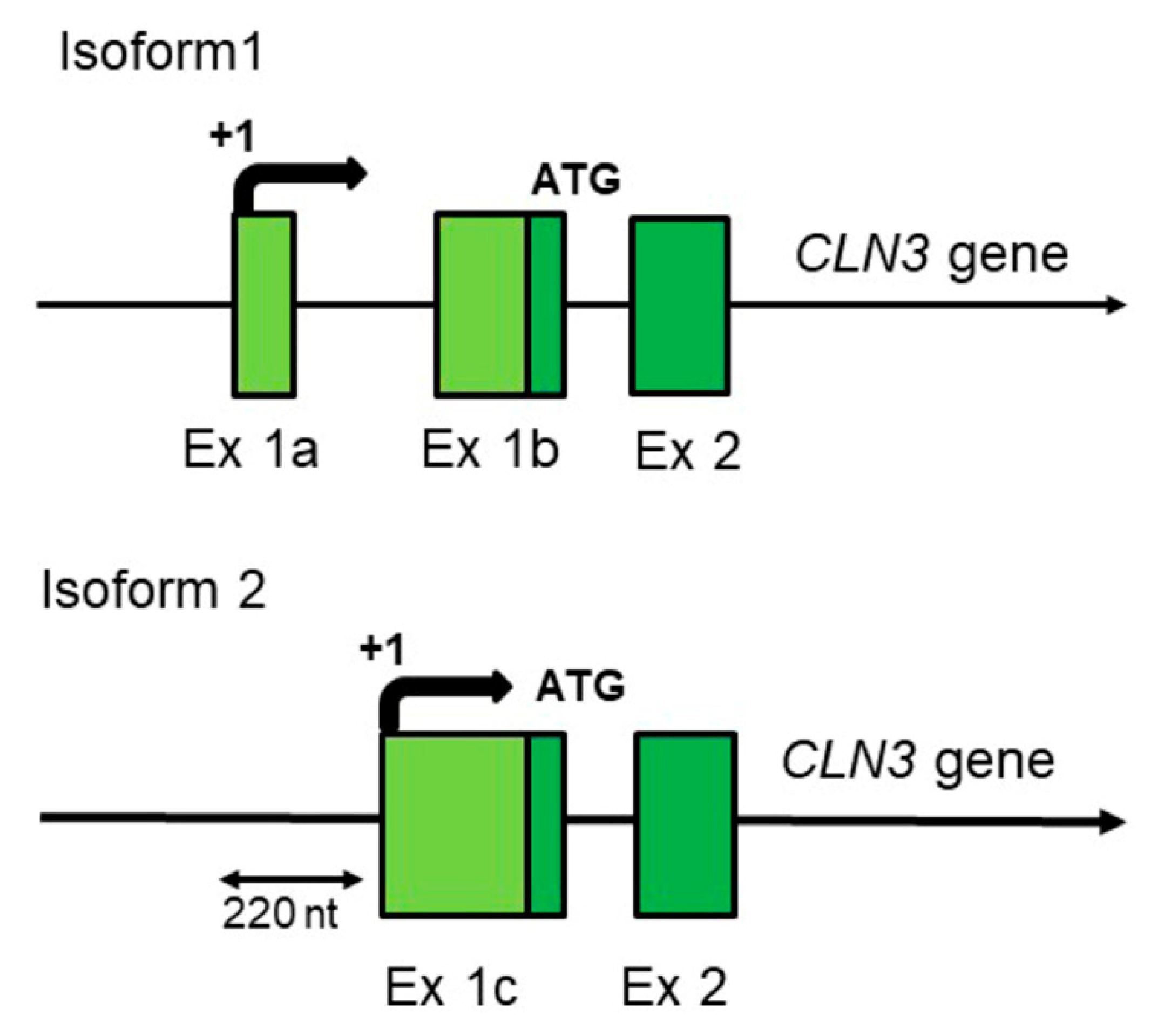
Although the CLN3 gene is highly conserved in evolution (from yeast to humans, and especially in mammals), genomic studies show that there are human-specific traits in its expression mechanisms. Indeed, as some other adaptively evolving genes [23], human CLN3 gene has two different transcription start points probably with very different regulation. The identical full-length hCLN3 protein can be synthesized from two different mRNAs transcribed from the same gene by using specific and alternatively regulated promoters (Figure 1).
Moreover, it is very likely that one of the transcripts (isoform 1) is generated by a specific form of RNA polymerase II containing minor isoform of the subunit hRPB11 (POLR2J), the product of expression of a human-specific POLR2J2 gene [24,25,26,27,28]. Recent identification of this (POLR2J2) gene as one of the only five identified biomarker genes for Batten disease [29] strongly argues in favor of this possibility.
To identify genetic variations responsible for the clinical variability of Batten disease, Lebrun et al. performed genome-wide microarray analyses in lymphocytes derived from patients with three different speed of the disease progression (slow, average and rapid). Five genes, called biomarkers, were found to be dysregulated by a similar way in all three patient groups: the expression of POLR2J2 and CDC42SE2 were decreased, whereas RGS1, DUSP2, and PARP15 were increased, in all CLN3 patients compared with controls.
The human specificity of the POLR2J2 and POLR2J3 genes was proven [24,25,27] and participation of products of their expression, minor subunits hRPB11bα (hRPB11cα) and hRPB11bβ (hRPB11cβ) of human RNA polymerase II, in formation of novel, human-specific transcription complexes was demonstrated in our previous works [28,30,31].
One of the surprising facts is that so far no large animals (dogs, cows, goats, etc.) have been found to suffer from a malady similar in symptoms to Batten disease (i.e., from the mutations or deletions in the CLN3 gene) [32]. To search for such animals, a separate project was created, which made it possible to detect large animals with mutations in almost all other CLN-genes discovered at that time [33,34,35]. In 2019, pigs were created that had a deletion in their CLN3 gene similar to that of patients with Batten disease (JNCL) [36]. However, at the moment there is still no information about what symptoms these animals show. For today the only mammalian species where the function of the CLN3 gene has been systematically addressed is mouse. Like its human counterpart, the mouse cDNA encodes a predicted 438-amino acid polypeptide which share 85% sequence identity with human protein [37]. Despite the obvious membrane deposition, Cln3−/− knock-in mice did not develop obvious clinical symptoms by 12 months of age [38]. And only in the second half of life, symptoms appear in a mild form: visual impairment, neuron loss, motor disfunctions, reduction of lifespan by 20% [39,40].
The presence of human-specific aspects of hCLN3 gene expression seems also related to the characteristic features of human neurons. In mice, monkeys, and other animals, the size of a neuron is limited by the level of expression of membrane proteins. At the same time, human cortical neurons are significantly larger and have a much larger number of dendrites. Moreover, in humans compared to all studied animals there is a drastic change in dendritic length with increasing radical distance from cell body [41]. In such specific cells as neurons, the limiting stage of their growth is the formation of giant membrane surfaces, and the CLN3 protein is undoubtedly involved in this process (discussed in [42]). It is quite possible that children in the period of active growth of their neurons need very effective production (high-performance synthesis) of battenin (CLN3 protein), which is achieved by the increased expression of the CLN3 mRNA isoform 1 (Figure 1), using a special form of RNA polymerase II containing the human-specific subunit POLR2J2 (hRPB11bα) in its composition (see Refs [28,30,31] for a discussion).
The isoform 1 CLN3 mRNA so far was predominantly isolated from the nervous tissue and thus may be at least to some extent brain-specific. In humans (at some stage of their development) this isoform is probably transcribed by the human-specific form of RNA polymerase II containing minor subunit POLR2J2 (hRPB11bα or hRPBbβ) [25,28,31].
Although originally, Batten disease was classified as one of the lysosomal storage disorders, to find the true causes of the disease, all other aspects of changes in cellular physiology need to be considered. Moreover, it is important to find a molecular explanation of how the well-characterized genetic defect (disruption of the CLN3 gene function) could affect cellular physiology of the neuronal tissue in a first place—the knowledge that is still largely missing. In the following sections of the review, we will sequentially consider specific physiological changes in cln3− cells (both from humans and from different model organisms, such as yeasts, slime mold, fruit fly and mouse) and discuss cellular systems and functions, primarily affected or damaged during progression of this neurodegenerative disease (Table 1).
3. The CLN3 Protein Mediates Anterograde and Retrograde Trafficking of Proteins and Other Substances
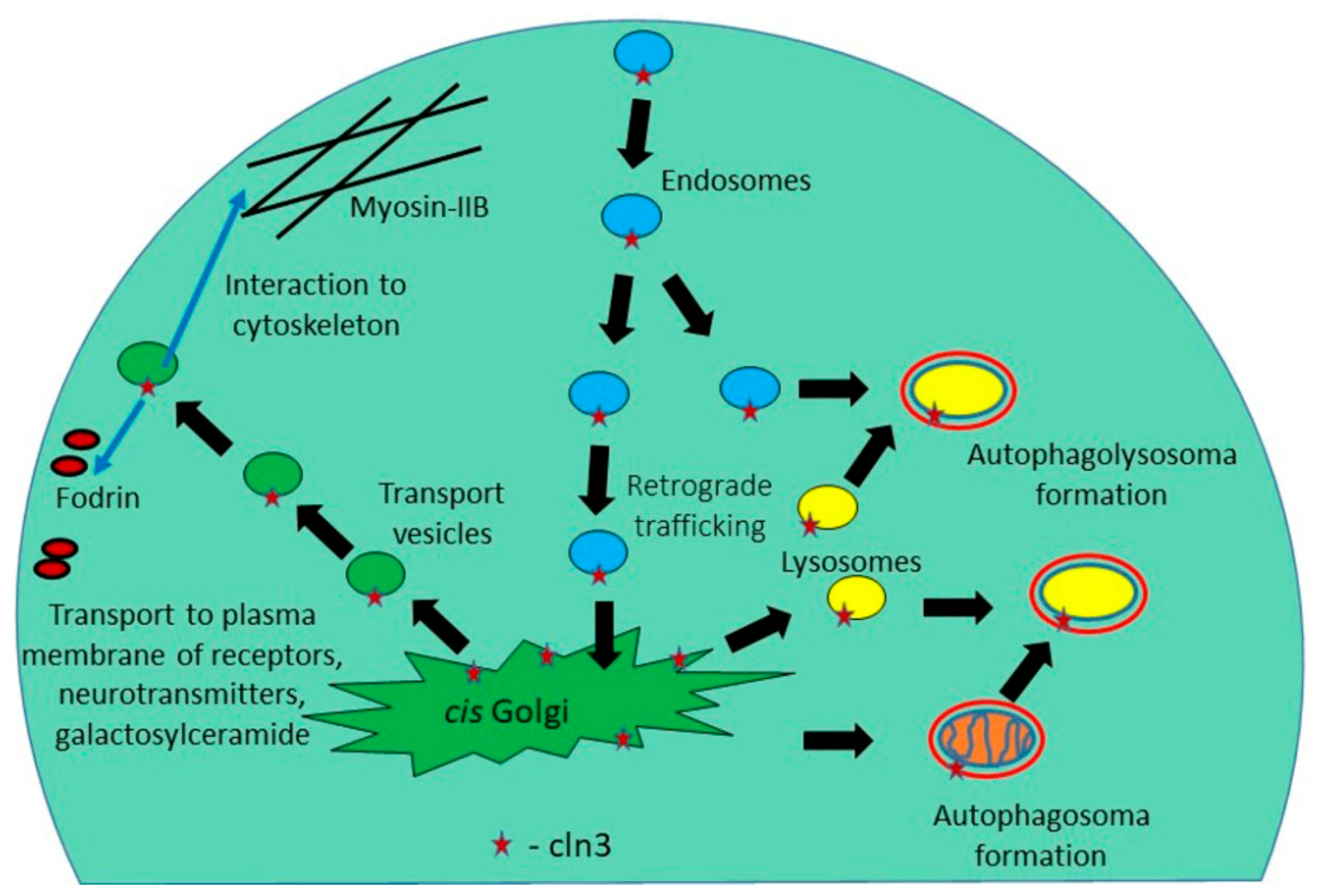
Production and distribution (delivery to destination) of СLN3 membrane protein is a rather complex and finely regulated process: СLN3 is synthesized in the endoplasmic reticulum, then transported to the Golgi apparatus and sent to the membranes of many cellular organelles (mitochondria, early and late endosomes and lysosomes) [43]. Since СLN3 is a component of the endosomal-lysosomal system, endocytosis [44], amino acid transport in vacuoles [45], autophagosomal maturation, as well as the stage of endosomal and lysosomal fusion are disturbed in the cells of JCNL patents [46]. Defects in the lysosomal-endosomal system lead to the formation of lysosomal inclusions, which have long been considered the cause of neurodegenerative processes occurring in patients suffering from Batten disease.
To identify components critical to lysosomal homeostasis that are affected by Batten disease, lysosomal proteomes were established in cerebellar cell lines derived from a Cln3-knock-in mouse model of human Batten disease and control cells. This analysis identified 70 proteins assigned to the lysosomal compartment and 3 lysosomal cargo receptors, which in most cases showed significant differential abundance between control and CLN3-defective cells. Among them, 28 soluble lysosomal proteins catalyzing the degradation of various macromolecules had decreased levels in CLN3-deficient cells. A decrease in the activity of 11 lipid-degrading lysosomal enzymes correlated with a reduced capability to degrade lipid droplets and some changes in the distribution and composition of membrane lipids. In particular, the levels of glycosphingolipids and lactosylceramides were decreased in CLN3-defective cells, which were also impaired in the exocyte transferrin receptor recirculation pathway [47]. Another signaling hub which includes two major mitogen-activated protein kinase (MAPK) cascades, and centers on the Tor kinase complexes TORC1 and TORC2, was identified in a global screen for genetic interactions with btn1, the fission yeast Schizosaccharomyces pombe orthologue of a human CLN3 gene [48]. In particular, it was shown that the TORC signaling pathway is repressed in yeast cells with deletion of the btn1 gene, which can also have a significant impact on the effectiveness of autophagy and lysosomogenesis.
Because Batten disease is primarily a neurodegenerative disorder, an important issue is the function of CLN3 membrane protein in brain tissue. In this regard, it is worth noting the work of Luiro and colleagues [49] who demonstrated that СLN3 is found in synaptosomal fractions isolated from nerve endings. This may involve interaction of CLN3 at the plasma membrane with components that regulate the actin cytoskeleton (β-fodrin and non-muscle myosin-IIB) [50,51] and/or with lipid rafts that are enriched in cholesterol and galactosylceramide [52,53]. The endocytic defect caused by CLN3 mutations can change neurotransmitter release and surface expression of receptors [49] and ion channels in neurons [72], leading to impaired neurotransmission and eventually neuronal cell death (Figure 2).
4. Stress of the Endoplasmic Reticulum (ER Stress)
Although trafficking of proteins is a pathway connecting the Golgi network, endosomes, autophagosomes, lysosomes and plasma membrane, it begins in the endoplasmic reticulum where the primary sorting of proteins occurs. A common feature of many neurodegenerative diseases is the accumulation of misfolded proteins that cause ER stress, affecting neuronal homeostasis and leading to cell death [54,73]. During times of ER stress, transcription of genes encoding secretory proteins is downregulated, misfolded proteins are cleared through ER-associated degradation (ERAD), more chaperones are synthesized in an attempt to increase the protein folding capacity. If the aforementioned mechanisms fail to rescue and restore cell homeostasis, then apoptosis could be triggered through the intrinsic pathway in an attempt to prevent further damage at the level of tissue or organism.
Eight NCL proteins can be connected in a single protein network [42]. Their interactions can occur only in the endoplasmic reticulum, so these proteins might have some ER-associated functions. Recently, it was shown that CLN8 is an endoplasmic reticulum cargo receptor [74] and CLN7 influences the secretion of Cln5 and CtsD in Dictyostelium discoideum [75].
To study the function of CLN3 in the ER-stress signaling pathway, Wu et al. compared proliferation and apoptosis in cells transfected by normal and mutant CLN3 after induction of ER stress with tunicamycin (TM). This drug blocks enzyme GlcNAc phosphotransferase (GPT), which catalyzes the transfer of N-acetylglucosamine-1-phosphate from UDP-N-acetylglucosamine to dolichol phosphate in the first step of glycoprotein synthesis. CLN3 protected cells from TM-induced apoptosis and increased cell proliferation. Increased expression of wild type CLN3 gene resulted in increased production of the ER chaperone protein GRP78 and decreased level of synthesis of proapoptotic protein CHOP. In contrast, increased expression of mutant cln3 or knockdown of CLN3 gene by siRNA had the opposite effect [55]. These findings suggest that СLN3 might be some kind of ER chaperone involved in the folding of glycoproteins, and its dysfunction in cells could lead to failure of the ER stress response program with switching on mechanism of apoptosis. That is why ER stress may be a key mechanism in the development of JNCL disease causing neuronal degeneration.
5. Dysfunction of Mitochondria
Mitochondrial dysfunction may also be one of the causes of neurodegeneration [56]. Morphological changes of mitochondria were observed in patients with JNCL: they are enlarged and lengthened [57]. Studies of skin fibroblasts from JNCL patients revealed a decrease in the basal activity of ATP synthase [58], the overall level of oxidative processes, a decrease in the concentration of AMP and energy-rich phosphates ATP and ADP [59]. In the mitochondria of JNCL patients, the rate of synthesis of fatty acids decreases, which leads to a mitochondrial membrane defect [60]. In addition, ATP synthase complexes function abnormally, since a large number of them are destroyed, and one of the subunits of this enzyme (namely, C) accumulates in lysosomal inclusions, which is one of the distinctive signs of Batten disease [61].
It was also shown that JNCL and other neuronal ceroid lipofuscinoses are characterized by selective loss of inhibitory GABAergic interneurons rich in mitochondria and are thus especially susceptible to compromised energy production [49]. Neuron loss primarily in brain areas most metabolically active and rich in mitochondria (cortical layers IV–V) in the JNCL patients also provides indirect evidence of mitochondrial involvement in the JNCL pathogenesis [62]. It was observed that genes of glycolysis (Gpi, Eno1, Tpi and Pfk1) are upregulated in embryonic primary culture of the Cln3−/− mouse neurons. Pfk1 (phosphofructokinase 1) is specific only to glycolysis, suggesting that upregulation of glycolysis may act as a compensatory mechanism for an inefficient energy production. Analysis of the mitochondrial function of the aforementioned Cln3-deficient mice also shows slightly reduced activities of the mitochondrial respiratory chain complexes, indicating reduced utilization of oxygen [49].
The negative effect on mitochondrial function in cln3− cells may be due to the suppression of the TORC1 gene function and the repression of TOR signaling, as observed in the cells of the fission yeast Schizosaccharomyces pombe with the deletion of the btn1 gene, a homologue of human CLN3 [48].
6. Reactive Oxygen Species (ROS)
An increase of reactive oxygen species (ROS) mediates different pathological processes such as atherosclerosis, diabetes, neurodegeneration, inflammation, and aging [76]. The observed alterations in macroautophagy can lead to an accumulation of oxidatively damaged mitochondria that in turn fuel the generation of ROS. Excess cellular levels of ROS cause damage to proteins, nucleic acids, lipids, membranes and organelles, which can lead to activation of cell death processes such as apoptosis. It was shown that ROS were significantly increased in cln3 deficient Batten disease patients fibroblasts in comparison with their appropriate controls [63]. Experiments with Drosophila melanogaster as a relatively simple model organism for studying Batten disease showed that the fruit fly cells lacking CLN3 are hypersensitive to oxidative stress yet they respond normally to other physiological stresses. Overexpression of the CLN3 gene is sufficient to confer increased resistance to oxidative stress. These data suggest that the lack of CLN3 function leads to a failure to manage the response to oxidative stress and this may be the key deficit in JNCL that leads to neuronal degeneration [64]. An increase of reactive oxygen species (ROS) can modify the cell-signaling proteins and have functional consequences, which mediate pathological processes neurodegeneration. As it was previously shown and reviewed, the generation and homeostasis of intracellular ROS could have impact on various cell-signaling (NF-κB, MAPKs, PI3K-Akt) and Ubiquitination/Proteasome systems, modifying protein kinases and ion channels and transporters (Ca2+ and mPTP) [77].
7. pH Homeostasis and Osmoregulation
Previously, it has shown that the BTN1 gene product of the yeast Saccharomyces cerevisiae is 39% identical and 59% similar to human CLN3, which is associated with the neurodegenerative disorder Batten disease. Yeast btn1-Δ strains have an elevated activity of the plasma membrane H+-ATPase due to an abnormally high vacuolar acidity during the early phase of growth [65].
Interesting results have been obtained with the social amoeba Dictyostelium discoideum, a soil microbe that has long been used as a model system for studying fundamental processes in cell biology and human neurological disorders, including NCLs. During starvation, Dictyostelium cells move from the stage of unicellular feeding, when cells grow and divide mitotically, to the stage of multicellular development. During development, individual cells aggregate and undergo a process of differentiation, forming a fruiting body, consisting of a stem of cells that support a mass of viable spores. In Dictyostelium discoideum, the CLN3 protein is localized primarily in the contractile vacuole (CV) system, which consists of a network of tubules that supply water to the bladders. During hypotonic stress, these bubbles of a social amoeba fill with water, and then periodically displace the accumulated water from the cell, collapsing on the plasma membrane. This mechanism allows cells to effectively regulate osmotic pressure in a variety of environments. It was shown that cln3− cells are sensitive to both hypotonic and hypertensive stress, which ultimately affects their viability and ability to multicellular development [66].
Hypertensive stress increased the expression of the CLN3 gene in the kidney cells of young hamsters and influenced the localization of the protein [67]. In addition, work on mice suggested a role for CLN3 in renal control of water and/or ionic homeostasis [68]. Finally, direct evidence for a role for CLN3 in osmoregulation was found when it was shown that regulatory volume depletion (RVD) is impaired in brain endothelial cells derived from CLN3-deficient mice [69]. RVD is a normal process that occurs when cells live in a hypotonic environment. The cells grow in size by absorbing excess water and then return to their normal size, displacing the water. Thus, if the process of osmoregulation is disturbed, the cell can be destroyed due to the inability to throw out excess water.
8. The Hyperactivation of Glial Cells and Astrocytes
Microglia are the resident immune cells within the central nervous system (CNS) parenchyma. The main functions of microglia are to phagocytose dead cells and release pro-inflammatory mediators. Abnormal neuron-microglia interactions have been implicated in the pathogenesis of several neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease [78]. Activated microglia are also observed in the brains of JNCL patients [70]. It was reported that microglia cells carrying the most common deletion of CLN3 gene (CLN3Δ ex7/8) exist in a pro-inflammatory state. Upon stimulation, microglia produce significantly more mediators of inflammation, including IL-1β, TNF-a, IL-1a, IL-9, and IL-15 [71]. cln3-deficient astrocytes are characterized by the reduced secretion of a range of neuroprotective factors, mitogens, chemokines and cytokines, in addition to impaired calcium signaling and glutamate clearance. Using a co-culture system, it was shown that cln3-deficient astrocytes and microglia had a negative impact on the survival and morphology of both cln3-deficient and wild type neurons. These data provide evidence that astrocytes in time of CLN3 disease are functionally compromised. Together with microglia, they may play an active role in neuron loss in this disorder and can be considered as potential targets for therapeutic interventions [71].
9. Unregulated Activation of Signal Cascades
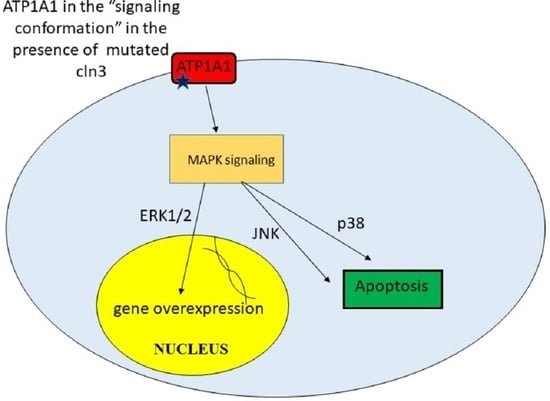
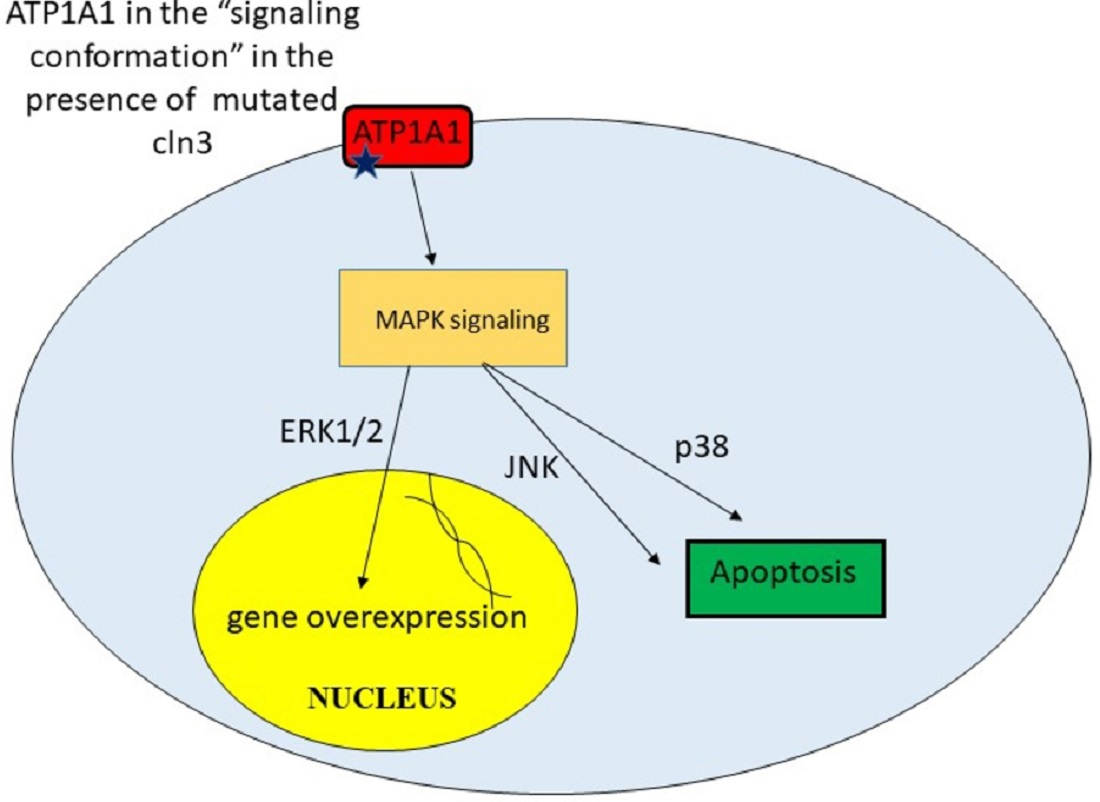
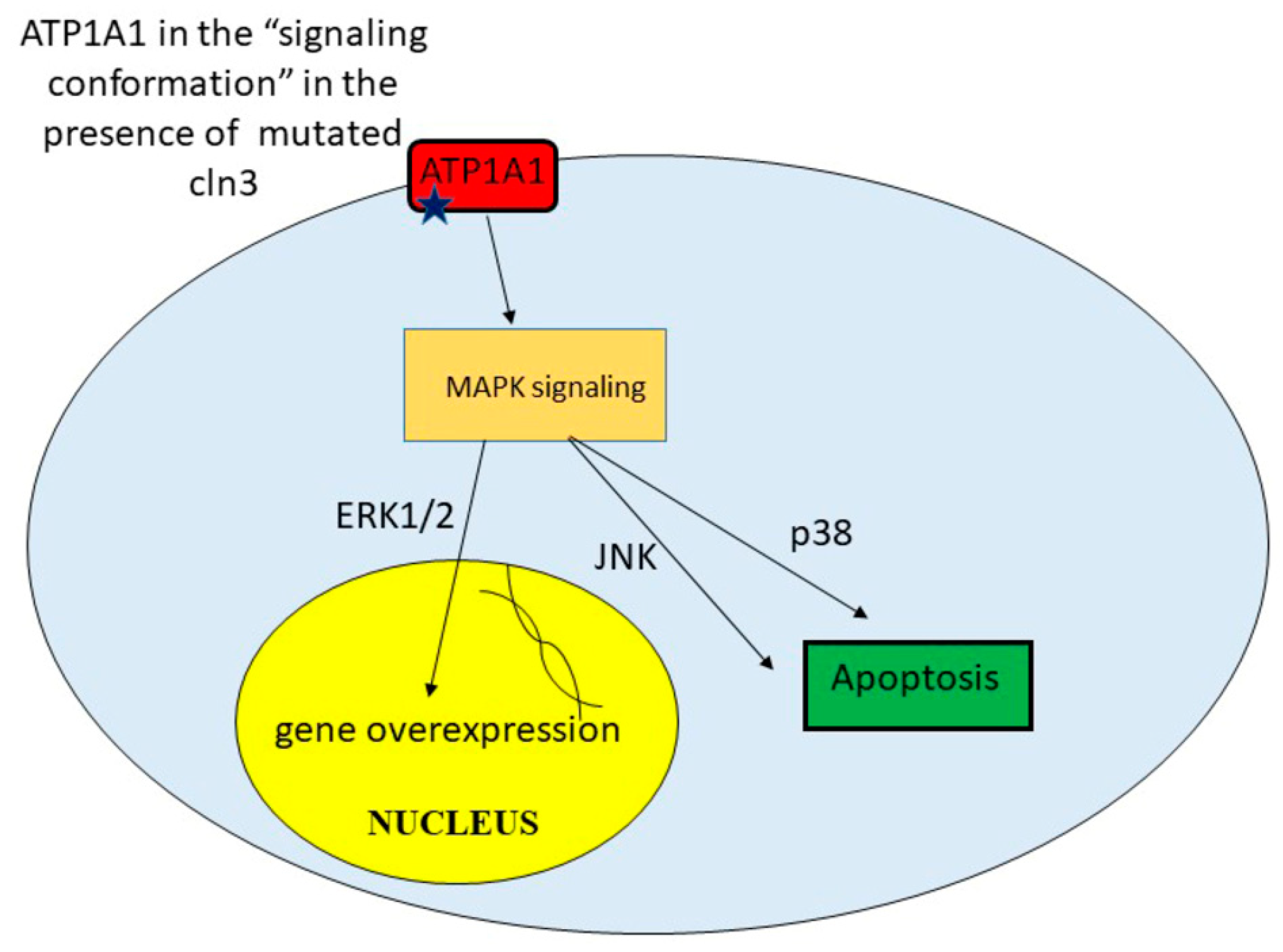
In Shematorova et al. [42], it was suggested that СLN3 is a chaperone of the endoplasmic reticulum and is involved in the formation of the three-dimensional structure of certain receptors, and its absence leads to changes in the 3D structure of these type of proteins. Incorrect folding of the plasma membrane proteins leads to initiation of signaling cascades from the cell surface. Although the true receptors that can trigger a signaling cascade from the membrane surface were not shown in the work, Uusi-Rauva et al. revealed that CLN3 interacts with ion channels such as Na+/K+ ATPase ATP1A1 (α1) subunit [50]. The α1 (first catalytic) subunit of Na+/K+ ATPase possesses both pumping and signaling functions. This protein, ATP1A1, might interact with non-receptor tyrosine kinase SRC and regulates its activity in a conformation-dependent manner [79]. It was also shown that cardiac glycoside ouabain [79], hydrogen peroxide [80], and ammonium chloride [81] might induce conformational changes in the tertiary structure of Na+/K+ ATPase ATP1A1 that leads to SRC and subsequent EGFR activation. So, conformational change in α1 Na+/K+ ATPase and its activation of SRC can be easily transmitted to MAPK cascades.
Indeed, recent studies have shown that SRC activation is required for ouabain-induced activation of ERK1/ERK2 and p38 [82]. MAPK (mitosis activated protein kinases) ERK1/ERK2 are responsible for cell growth and proliferation and are involved in the activation of genes that provide synthesis of growth factors and mitogens. However, excessive uncontrolled activation of these signaling pathways can lead to the overproduction of certain proteins and their toxicity, which can lead to cell death. The p38 and JUN pathways have also been implicated in stress-induced signaling leading to apoptosis [83]. Thus, CLN3 may be involved in the correct formation of the spatial structure of ATP1A1 in neurons. Mutations or truncations of this protein lead to a shift in ATP1A1 signaling and activation of EGFR via SRC kinase [83] (Figure 3).
Recently by using comparative transcriptomics Huber and Mathavarajah identified genes whose expression is significantly increased in the social amoeba Dictyostelium discoideum cln3− cells during starvation. In these conditions Dictyostelium cells secrete cAMP, which acts as a chemoattractant causing cells to aggregate into multicellular mounds and after that in the multicellular fruitful body, forming spores [84]. cln3−cells displayed delayed migration and aggregation that support aberrant signaling during this stage of development [83]. Expression of a number of genes encoding MAPK signaling cascade proteins are elevated under starvation conditions in Dictyostelium cln3− cells, which indicates the importance of CLN3 for proper functioning of this particular regulatory pathway [85].
10. Current Treatment Opportunities
Perhaps the most promising method of treating JNCL today is the introduction of the normal CLN3 gene into the spinal cord of patients using adeno-associated virus (AAV) constructs [86,87]. Indeed, AAV-mediated gene therapies have proven as a successful treatment strategy to combat photoreceptor degenerations. Kleine Holthaus et al. developed a successful treatment for the retinal degeneration in Cln6-deficient mice, a model of NCL that carries a mutation in the Cln6 gene [88]. When the therapeutic CLN6 transgene was delivered specifically to bipolar cells, a retinal cell type downstream of photoreceptors, degeneration of photoreceptor cells became significantly less strong [89].
Another important breakthrough has been the application of enzyme replacement technique (Bruniera) for CLN2 disease, the first ever FDA-approved therapy, which is significantly improving quality of life and delaying disease progression for persons affected by this disease [90]. Unfortunately, this type of “cross-correction” which is applicable for enzyme deficient forms of NCL could not be used in case of Batten disease.
At the moment, phase I/IIa clinical trials of gene therapy are currently in progress for two other forms of NCL (CLN3 disease: NCT0377057233 and CLN6 disease: NCT0272558034). In case of CLN3 (Batten) disease, recently initiated by Amicus Therapeutics treatment consists of a one-time low or high dose injection of AT-GTX-502 construct (scAAV9.P546.CLN3) into the lumbar spinal cord region of subjects with CLN3 Batten disease and their monitoring over a period of at least three years [91].
Concerning the examples of gene therapy approaches mentioned above, the main issues are the presence of naturally occurring antibodies against adeno-associated viruses (AAVs) in a variable proportion of the patient population and the open question whether transgene expression will be sustained for a long period of time — re-administration of vectors right now is hardly possible [92].
Another way of treatment is to use a splice-modulating antisense oligonucleotides drug tailored to a particular patient [93]. There is no serious adverse events, and treatment leads to objective reduction in seizures (determined by electroencephalography and parental reporting). However, this method can only be used for patients with proven aberrant splicing in the synthesis of CLN3 mRNA, as a result of which the correct reading frame of the battenin (CLN3 protein) cannot be formed.
Using mouse models, drugs have been discovered that fight certain cell disorders caused by CLN3 deficiency, such as neuroinflammation [94,95], imposed autophagy and reduced mitochondrial membrane potential [96], apoptosis [97]. Mycophenolate, an immunosuppressant, today is already used in the treatment of patients with Batten disease [98].
Unfortunately, all the methods listed above are universal for the treatment of all monogenic recessive hereditary diseases and do not rely on understanding the mechanisms of development of a particular disease.
11. Conclusions
Batten disease or JNCL is an incurable devastating disease affecting primarily young children. Currently, there is no curative therapy for this malady, and all available treatments for JNCL are symptomatic and palliative. Although originally Batten disease was classified as one of the lysosomal storage diseases and many studies were aimed at investigating this particular aspect of the malady, these studies did not lead to understanding of the molecular mechanisms of this disease and/or finding potential ways to treat it. The main mechanisms of Batten disease as a neurodegenerative disorder can be associated with many other defects in cellular physiology summarized and discussed in the present review.
As noted earlier, all animal models of JNCL disease studied so far do not exhibit human-specific symptoms. For example, the introduction of 1.02-kb deletion in the mouse cln3 gene causes the retina degeneration only in the second half of the animals’ life, and all the other symptoms are manifested in a mild form [32]. Therefore, it is not surprising that at the moment only palliative care is available for patients with Batten disease. It is possible that only gene therapy using adenovirus-based vectors can bring some positive effect(s), as was observed in the case of enzyme replacement therapy for Jansky-Bielschowsky disease (NCL2) caused by mutations in the CLN2 gene [90].
It is likely that the disease process may involve a special battenin-dependent pathway that is essential for neuronal cell survival, and thus consistent with battenin-specific function in neurons. In connection with the discovery of human-specific features of CLN3 gene expression, it is important to study in detail both promoters of this gene and the expression of two isoforms of its full-size mRNA in different tissues, as well as the possible participation of human-specific genes POLR2J2 (and POLR2J3) in this process. This may help, in particular, to clarify the interesting fact noted by some researchers that although the level of CLN3 mRNA in the brain is low compared to other tissues (please, see Transcriptome Databases), it is in the cells of the nervous tissue that the extremely high (the highest) level of synthesis of the battenin (CLN3 protein) is observed [99]. A thorough understanding of the mechanisms regulating disease progression can help determine new treatments for Batten disease. Without an understanding of the molecular mechanisms of Batten disease, it will be impossible to fight the illness.
Authors Contributions
E.K.S. conceived the work, designed and drafted the manuscript; G.V.S. conceived the work, revised the manuscript and supervised the work. Both authors have read and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The study was carried out under governmental support (salary and facility maintenance) of the scientific project No. 01201352888 (the task No. 0101-2014-0042) and was supported in part by the grant No. 14-04-01485 [2014–2016] from Russian Foundation for Basic Research [RFBR] (to G.V.S.). Publication costs of the article were covered by the grant from the RFBR No. 18-04-01262 [2018–2020] (to G.V.S.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ER | Endoplasmic Reticulum |
| JNCL | Juvenile Neuronal Ceroid Lipofuscinosis |
| NCLs | Neuronal Ceroid Lipofuscinoses |
| mPTP | Mitochondrial Permeability Transition Pore |
| ROS | Reactive Oxygen Species |
References
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.; Vainshtein, A.; di Ronza, A.; Chandrachud, U.; Haslett, L.J.; Palmieri, M.; Storch, S.; Groh, J.; Dobzinski, N.; Napolitano, G.; et al. The CLN3 gene and protein: What we know. Mol. Genet. Genom. Med. 2019, 7, e859. [Google Scholar] [CrossRef]
- Marshall, F.J.; de Blieck, E.A.; Mink, J.W.; Dure, L.; Adams, H.; Messing, S.; Rothberg, P.G.; Levy, E.; McDonough, T.; Deyoung, J.; et al. A clinical rating scale for Batten disease: Reliable and relevant for clinical trials. Neurolgy 2005, 65, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Collins, J. Batten disease: Features to facilitate early diagnosis. Br. J. Ophthalmol. 2006, 90, 1119–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouseph, M.M.; Kleinman, M.E.; Wang, Q.J. Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Ann. New York Acad. Sci. 2016, 1371, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, A.K.; Drack, A.V.; Ostergaard, J.R. Cataract and Glaucoma Development in Juvenile Neuronal Ceroid Lipofuscinosis (Batten Disease). Ophthalmic Genet. 2014, 36, 39–42. [Google Scholar] [CrossRef]
- Adams, H.R.; Mink, J.W.; University of Rochester Batten Center Study Group. Neurobehavioral Features and Natural History of Juvenile Neuronal Ceroid Lipofuscinosis (Batten Disease). J. Child Neurol. 2013, 28, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.M.; Adams, H.; Rothberg, P.G.; Augustine, E.F.; Marshall, F.J.; Deblieck, E.A.; Vierhile, A.; Beck, C.A.; Newhouse, N.J.; Cialone, J.; et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011, 77, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claussen, M.; Heim, P.; Knispel, J.; Goebel, H.H.; Kohlschütter, A. Incidence of neuronal ceroid-lipofuscinoses in West Germany: Variation of a method for studying autosomal recessive disorders. Am. J. Med. Genet. 1992, 42, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, H.M.; O’Rawe, A.M.; Taschner, P.E.; Sandkuijl, L.A.; Santavuori, P.; de Vos, N.; Breuning, M.H.; Mole, S.E.; Gardiner, R.M.; Järvelä, I.E. Batten disease gene, CLN3: Linkage disequilibrium mapping in the Finnish population, and analysis of European haplotypes. Am. J. Hum. Genet. 1995, 56, 654–662. [Google Scholar] [PubMed]
- Munroe, P.B.; Mitchison, H.M.; O’Rawe, A.M.; Anderson, J.W.; Boustany, R.-M.; Lerner, T.J.; Taschner, P.E.; de Vos, N.; Breuning, M.H.; Gardiner, R.M.; et al. Spectrum of Mutations in the Batten Disease Gene, CLN3. Am. J. Hum. Genet. 1997, 61, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Jarvela, I.; Mitchison, H.M.; Munroe, P.B.; O’Rawe, A.M.; Mole, S.E.; Syvanen, A.C. Rapid diagnostic test for the major mutation underlying Batten disease. J. Med. Genet. 1996, 33, 1041–1042. [Google Scholar] [CrossRef] [Green Version]
- Jilani, A.; Matviychuk, D.; Blaser, S.; Dyack, S.; Mathieu, J.; Prasad, A.N.; Prasad, C.; Kyriakopoulou, L.; Mercimek-Andrews, S. High diagnostic yield of direct Sanger sequencing in the diagnosis of neuronal ceroid lipofuscinoses. JIMD Rep. 2019, 50, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Rietdorf, K.; Coode, E.E.; Schulz, A.; Wibbeler, E.; Bootman, M.D.; Ostergaard, J.R. Cardiac pathology in neuronal ceroid lipofuscinoses (NCL): More than a mere co-morbidity. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165643. [Google Scholar] [CrossRef]
- Radke, J.; Koll, R.; Gill, E.; Wiese, L.; Schulz, A.; Kohlschütter, A.; Schuelke, M.; Hagel, C.; Stenzel, W.; Goebel, H.H. Autophagic vacuolar myopathy is a common feature of CLN3 disease. Ann. Clin. Transl. Neurol. 2018, 5, 1385–1393. [Google Scholar] [CrossRef]
- Lerner, T.J.; Boustany, R.-M.N.; Anderson, J.W.; D’Arigo, K.L.; Schlumpf, K.; Buckler, A.J.; Gusella, J.F.; Haines, J.L. Isolation of a novel gene underlying batten disease, CLN3. Cell 1995, 82, 949–957. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A. The mutational constraint spectrum quantified from variation in 141,456 humans. Nat. Cell Biol. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kitzmüller, C.; Haines, R.L.; Codlin, S.; Cutler, D.F.; Mole, S.E. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2007, 17, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, R.W.; Munroe, P.B.; Mitchison, H.M.; Gardiner, R.M.; Mole, S.E.; Wallace, B.A. A model for Batten disease protein CLN3: Functional implications from homology and mutations. FEBS Lett. 1996, 399, 75–77. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.-I.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2005, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Grandemange, S.; Schaller, S.; Yamano, S.; du Manoir, S.; Shpakovski, G.V.; Mattei, M.-G.; Kedinger, C.; Vigneron, M. A human RNA polymerase II subunit is encoded by a recently generated multigene family. BMC Mol. Biol. 2001, 2, 14. [Google Scholar] [CrossRef]
- Shpakovski, D.G.; Shematorova, E.K.; Shpakovski, G.V. New Genes on Human Chromosome 7: Bioinformation Analysis of a Cluster of Genes from the POLR2J Family. Russ. J. Bioorganic Chem. 2004, 30, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Benga, W.J.; Grandemange, S.; Shpakovski, G.V.; Shematorova, E.K.; Kedinger, C.; Vigneron, M. Distinct regions of RPB11 are required for heterodimerization with RPB3 in human and yeast RNA polymerase II. Nucleic Acids Res. 2005, 33, 3582–3590. [Google Scholar] [CrossRef] [Green Version]
- Shematorova, E.K.; Shpakovski, D.G.; Shpakovski, G.V. PMS2 and POLR2J gene families as molecular markers of the higher primate’s evolution. Russ. J. Genet. 2010, 46, 1112–1114. [Google Scholar] [CrossRef]
- Shematorova, E.K.; Shpakovski, D.G.; Shpakovski, G.V. Novel complexes of gene expression and their role in the appearance and evolution of the genus Homo. Cell Tissue Biol. 2013, 7, 314–319. [Google Scholar] [CrossRef]
- Lebrun, A.-H.; Moll-Khosrawi, P.; Pohl, S.; Makrypidi, G.; Storch, S.; Kilian, D.; Streichert, T.; Otto, B.; Mole, S.E.; Ullrich, K.; et al. Analysis of Potential Biomarkers and Modifier Genes Affecting the Clinical Course of CLN3 Disease. Mol. Med. 2011, 17, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Proshkin, S.A.; Shematorova, E.K.; Souslova, E.A.; Proshkina, G.M.; Shpakovski, G.V. A minor isoform of the human RNA polymerase II subunit hRPB11 (POLR2J) interacts with several components of the translation initiation factor eIF3. Biochemistry 2011, 76, 976–980. [Google Scholar] [CrossRef]
- Proshkin, S.A.; Shematorova, E.K.; Shpakovski, G.V. The Human Isoform of RNA Polymerase II Subunit hRPB11bα Specifically Interacts with Transcription Factor ATF4. Int. J. Mol. Sci. 2019, 21, 135. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Hughes, S.M.; Liu, W.; Morgan, A.; Tuxworth, R.I.; Russell, C. The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165614. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.D.; Martinus, R.D.; Palmer, D.N. Sheep and other animals with ceroid-lipofuscinoses: Their relevance to Batten disease. Am. J. Med Genet. 1992, 42, 609–614. [Google Scholar] [CrossRef]
- Koppang, N. English setter model and juvenile ceroid-lipofuscinosis in man. Am. J. Med Genet. 1992, 42, 599–604. [Google Scholar] [CrossRef]
- Taylor, R.M.; Farrow, B.R.H. Ceroid lipofuscinosis in the Border Collie dog: Retinal lesions in an animal model of juvenile Batten disease. Am. J. Med Genet. 1992, 42, 622–627. [Google Scholar] [CrossRef]
- Johnson, T.B.; Sturdevant, D.A.; White, K.A.; Drack, A.V.; Bhattarai, S.; Rogers, C.; Cooper, J.D.; Pearce, D.A.; Weimer, J.M. Characterization of a novel porcine model of CLN3-Batten disease. Mol. Genet. Metab. 2019, 126, S81. [Google Scholar] [CrossRef]
- Lee, R.L.; Johnson, K.R.; Lerner, T.J. Isolation and Chromosomal Mapping of a Mouse Homolog of the Batten Disease GeneCLN3. Genomics 1996, 35, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Vrbanac, V.; Lebel, L.-A.; Lee, R.L.; Johnson, K.A.; Donahue, L.-R.; Teed, A.M.; Antonellis, K.; Bronson, R.T.; Lerner, T.J.; et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 2002, 11, 2709–2721. [Google Scholar] [CrossRef]
- Katz, M.L.; Shibuya, H.; Liu, P.C.; Kaur, S.; Gao, C.L.; Johnson, G.S. A mouse gene knockout model for juvenile ceroid-lipofuscinosis (Batten disease). J. Neurosci. Res. 1999, 57, 551–556. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Bernard, D.J.; Greene, N.D.E.; Cooper, J.D.; Junaid, M.A.; Pullarkat, R.K.; de Vos, N.; Breuning, M.H.; Owens, J.W.; Mobley, W.C.; et al. Targeted Disruption of the Cln3 Gene Provides a Mouse Model for Batten Disease. Neurobiol. Dis. 1999, 6, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.; Verhoog, M.B.; Doreswamy, K.K.; Eyal, G.; Aardse, R.; Lodder, B.N.; Goriounova, N.A.; Asamoah, B.; Brakspear, A.C.B.; Groot, C.; et al. Dendritic and Axonal Architecture of Individual Pyramidal Neurons across Layers of Adult Human Neocortex. Cereb. Cortex 2015, 25, 4839–4853. [Google Scholar] [CrossRef] [PubMed]
- Shematorova, E.K.; Shpakovski, D.G.; Chernysheva, A.D.; Shpakovski, G.V. Molecular mechanisms of the juvenile form of Batten disease: Important role of MAPK signaling pathways (ERK1/ERK2, JNK and p38) in pathogenesis of the malady. Biol. Direct. 2018, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kida, E.; Kaczmarski, W.; Golabek, A.; Kaczmarski, A.; Michalewski, M.; Wisniewski, K. Analysis of Intracellular Distribution and Trafficking of the CLN3 Protein in Fusion with the Green Fluorescent Proteinin Vitro. Mol. Genet. Metab. 1999, 66, 265–271. [Google Scholar] [CrossRef]
- Fossale, E.; Wolf, P.; Espinola, J.A.; Lubicz-Nawrocka, T.; Teed, A.M.; Gao, H.; Rigamonti, D.; Cattaneo, E.; MacDonald, M.E.; Cotman, S.L. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Ramirez-Montealegre, D.; Pearce, D.A. A role in vacuolar arginine transport for yeast Btn1p and for human CLN3, the protein defective in Batten disease. Proc. Natl. Acad. Sci. USA 2003, 100, 15458–15462. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; Macdonald, M.E.; Cotman, S.L. Autophagy Is Disrupted in a Knock-in Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493. [Google Scholar] [CrossRef] [Green Version]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Zheng, H.; Zhao, C.; Soherwardy, A.; Moore, D.F.; Lobel, P. Analysis of Brain and Cerebrospinal Fluid from Mouse Models of the Three Major Forms of Neuronal Ceroid Lipofuscinosis Reveals Changes in the Lysosomal Proteome. Mol. Cell. Proteom. 2019, 18, 2244–2261. [Google Scholar] [CrossRef]
- Bond, M.E.; Brown, R.; Rallis, C.; Bähler, J.; Mole, S.E. A central role for TOR signalling in a yeast model for juvenile CLN3 disease. Microb. Cell 2015, 2, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Luiro, K.; Kopra, O.; Lehtovirta, M.; Jalanko, A. CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: New clues to Batten disease. Hum. Mol. Genet. 2001, 10, 2123–2131. [Google Scholar] [CrossRef] [Green Version]
- Uusi-Rauva, K.; Luiro, K.; Tanhuanpää, K.; Kopra, O.; Martín-Vasallo, P.; Kyttälä, A.; Jalanko, A. Novel interactions of CLN3 protein link batten disease to dysregulation of fodrin-Na+, K+ ATPase complex. Exp Cell Res. 2008, 314, 2895–2905. [Google Scholar] [CrossRef]
- Getty, A.; Benedict, J.; Pearce, D. A novel interaction of CLN3 with nonmuscle myosin-IIB and defects in cell motility of Cln3−/− cells. Exp. Cell Res. 2011, 317, 51–69. [Google Scholar] [CrossRef] [Green Version]
- Persaud-Sawin, D.-A.; McNamara, J.O.; Rylova, S.N.; Vandongen, A.M.J.; Boustany, R.-M.N. A Galactosylceramide Binding Domain Is Involved in Trafficking of CLN3 from Golgi to Rafts via Recycling Endosomes. Pediatr. Res. 2004, 56, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Narayan, S.B.; Pastor, J.V.; Bennett, M.J. CLN3P, the Batten disease protein, localizes to membrane lipid rafts (detergent-resistant membranes). Biochem. Biophys. Res. Commun. 2004, 317, 988–991. [Google Scholar] [CrossRef]
- Marotta, D.; Tinelli, E.; Mole, S.E. NCLs and ER: A stressful relationship. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1273–1281. [Google Scholar] [CrossRef]
- Wu, D.; Liu, J.; Wu, B.; Tu, B.; Zhu, W.-G.; Luo, J. The Batten disease gene CLN3 confers resistance to endoplasmic reticulum stress induced by tunicamycin. Biochem. Biophys. Res. Commun. 2014, 447, 115–120. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zeman, W.; Donahue, S. Fine structure of the lipid bodies in juvenile amaurotic idiocy. Acta Neuropathol. 1963, 3, 144–149. [Google Scholar] [CrossRef]
- Das, A.; Jolly, R.; Kohlschütter, A. Anomalies of Mitochondrial ATP Synthase Regulation in Four Different Types of Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 1999, 66, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; von Harlem, R.; Feist, M.; Lücke, T.; Kohlschütter, A.; Kohlschütter, A. Altered levels of high-energy phosphate compoundsin fibroblasts from different forms of neuronal ceroid lipofuscinoses: Further evidence for mitochondria) involvement. Eur. J. Paediatr. Neurol. 2001, 5, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Kilkus, J.; Siakotos, A.N.; Singh, I. Mitochondrial abnormalities in CLN2 and CLN3 forms of batten disease. Mol. Chem. Neuropathol. 1996, 29, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP synthase subunitc storage in the ceroid-lipofuscinoses (Batten disease). Am. J. Med Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Goebel, H.H. Loss of pigment-laden stellate cells: A severe alteration of the isocortex in juvenile neuronal ceroid-lipofuscinosis. Acta Neuropathol. 1978, 42, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Donet, J.M.; Cárcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS Activity and Lysosomal pH Account for Distinct Patterns of Macroautophagy in LINCL and JNCL Fibroblasts. PLoS ONE 2013, 8, e55526. [Google Scholar] [CrossRef] [Green Version]
- Tuxworth, R.I.; Chen, H.; Vivancos, V.; Carvajal, N.; Huang, X.; Tear, G. The Batten disease gene CLN3 is required for the response to oxidative stress. Hum. Mol. Genet. 2011, 20, 2037–2047. [Google Scholar] [CrossRef] [Green Version]
- Padilla-Lopez, S.; Pearce, D.A. Saccharomyces cerevisiae Lacking Btn1p Modulate Vacuolar ATPase Activity to Regulate pH Imbalance in the Vacuole. J. Biol. Chem. 2006, 281, 10273–10280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathavarajah, S.; McLaren, M.D.; Huber, R.J. Cln3 function is linked to osmoregulation in a Dictyostelium model of Batten disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3573. [Google Scholar] [CrossRef] [PubMed]
- Getty, A.; Kovács, A.D.; Lengyel-Nelson, T.; Cardillo, A.; Hof, C.; Chan, C.-H.; Pearce, D.A. Osmotic Stress Changes the Expression and Subcellular Localization of the Batten Disease Protein CLN3. PLoS ONE 2013, 8, e66203. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.S.; Yancey, P.H.; Martins, I.; Sigmund, R.D.; Stokes, J.B.; Davidson, B.L. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am. J. Physiol. Physiol. 2010, 298, C1388–C1400. [Google Scholar] [CrossRef]
- Tecedor, L.; Stein, C.S.; Schultz, M.L.; Farwanah, H.; Sandhoff, K.; Davidson, B.L. CLN3 Loss Disturbs Membrane Microdomain Properties and Protein Transport in Brain Endothelial Cells. J. Neurosci. 2013, 33, 18065–18079. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Kielian, T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype. J. Neurochem. 2013, 127, 245–258. [Google Scholar] [CrossRef]
- Lange, J.; Haslett, L.J.; Lloyd-Evans, E.; Pocock, J.M.; Sands, M.S.; Williams, B.P.; Cooper, J.D. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2018, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-W.; Choi, H.; Kim, H.-J.; Jo, D.-G.; Jeon, Y.-J.; Noh, J.-Y.; Park, W.J.; Jung, Y.-K. Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin. Hum. Mol. Genet. 2006, 16, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Cabral-Miranda, F.; Hetz, C. ER Stress and neurodegenerative disease: A cause or effect relationship? Curr. Top Microbiol. Immunol. 2018, 414, 131–157. [Google Scholar]
- di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S.; Yap, S.Q. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell. Signal. 2020, 70, 109572. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Pheromone Signal. 2019, 2034, 57–67. [Google Scholar] [CrossRef]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol Cell. 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D. Ammonium Activates Ouabain-Activated Signalling Pathway in Astrocytes: Therapeutic Potential of Ouabain Antagonist. Curr. Neuropharmacol. 2014, 12, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.L.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Mathavarajah, S. Comparative transcriptomics reveals mechanisms underlying cln3-deficiency phenotypes in Dictyostelium. Cell. Signal. 2019, 58, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Loss of Cln3 impacts protein secretion in the social amoeba Dictyostelium. Cell. Signal. 2017, 35, 61–72. [Google Scholar] [CrossRef]
- Cooper, J.D.; Mole, S.E. Future perspectives: What lies ahead for Neuronal Ceroid Lipofuscinosis research? Biochim. Biophys. Acta Mol. Basis Dis. 2020, 8, 165681. [Google Scholar] [CrossRef]
- Liu, W.; Kleine-Holthaus, S.-M.; Herranz-Martin, S.; Aristorena, M.; Mole, S.E.; Smith, A.J.; Ali, R.R.; Rahim, A.A. Experimental gene therapies for the NCLs. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165772. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Herranz-Martin, S.; Massaro, G.; Aristorena, M.; Hoke, J.; Hughes, M.P.; Maswood, R.; Semenyuk, O.; Basche, M.; Shah, A.Z.; et al. Neonatal brain-directed gene therapy rescues a mouse model of neurodegenerative CLN6 Batten disease. Hum. Mol. Genet. 2019, 28, 3867–3879. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Ribeiro, J.; Abelleira-Hervas, L.; Pearson, R.A.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; Rizzi, M.; Hoke, J.; Maswood, R.; et al. Prevention of Photoreceptor Cell Loss in a Cln6 Mouse Model of Batten Disease Requires CLN6 Gene Transfer to Bipolar Cells. Mol. Ther. 2018, 26, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Ajayi, T.; Specchio, N.; Reyes, E.D.L.; Gissen, P.; Ballon, D.J.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. New Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- NCT03770572: Gene Transfer Study of AAV9-CLN3 for Treatment NCL Type 3. Available online: https://clinicaltrials.gov/ct2/show/NCT03770572?cond=CLN3&rank=1 (accessed on 22 November 2019).
- Berns, K.I.; Muzyczka, N. AAV: An Overview of Unanswered Questions. Hum. Gene Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Hu, C.; el Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. New Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Tarczyluk-Wells, M.A.; Salzlechner, C.; Najafi, A.R.; Lim, M.J.; Smith, D.; Platt, F.M.; Williams, B.P.; Cooper, J.D. Combined Anti-inflammatory and Neuroprotective Treatments Have the Potential to Impact Disease Phenotypes in Cln3−/− Mice. Front. Neurol. 2019, 10, 963. [Google Scholar] [CrossRef] [Green Version]
- Groh, J.; Berve, K.; Martini, R. Fingolimod and Teriflunomide Attenuate Neurodegeneration in Mouse Models of Neuronal Ceroid Lipofuscinosis. Mol. Ther. 2017, 25, 1889–1899. [Google Scholar] [CrossRef] [Green Version]
- Makoukji, J.; Saadeh, F.; Mansour, K.A.; Al Ali, J.; Kinarivala, N.; Trippier, P.C.; El-Sitt, S.; Boustany, R.-M. Flupirtine derivatives as potential treatment for the neuronal ceroid lipofuscinoses. Ann. Clin. Transl. Neurol. 2018, 5, 1089–1103. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Song, K.D.; Lee, H.-K.; Yi, S.; Lee, Y.S.; Heo, T.-H.; Jun, H.S.; Kim, S.-J. Fibrates inhibit the apoptosis of Batten disease lymphoblast cells via autophagy recovery and regulation of mitochondrial membrane potential. Vitr. Cell. Dev. Biol. Anim. 2015, 52, 349–355. [Google Scholar] [CrossRef]
- Augustine, E.F.; Beck, C.A.; Adams, H.R.; Defendorf, S.; Vierhile, A.; Timm, D.; Weimer, J.M.; Mink, J.W.; Marshall, F.J. Short-Term Administration of Mycophenolate Is Well-Tolerated in CLN3 Disease (Juvenile Neuronal Ceroid Lipofuscinosis). JIMD Rep. 2018, 43, 117–124. [Google Scholar] [CrossRef]
- Margraf, L.R.; Boriack, R.L.; Routheut, A.A.; Cuppen, I.; AlHilali, L.; Bennett, C.J.; Bennett, M.J. Tissue Expression and Subcellular Localization of CLN3, the Batten Disease Protein. Mol. Genet. Metab. 1999, 66, 283–289. [Google Scholar] [CrossRef]
Figure 1.
The schematic structure of two different full-length (complete) CLN3 mRNA isoforms transcribed from varying promoters of the CLN3 gene of Homo sapiens. Only regions corresponding to distinguished (differing) transcription start points and 2–3 of the first exons (out of 15–17) of the hCLN3 gene are shown. hCLN3 isoform 1 (transcript variant 1)—3832 nt (GenBank NM_001042432, UniProtKB/Swiss-Prot Q13286, CCDS 10632, Ensembl-Tr: ENST00000636147 (transcript: CLN3-244), protein—ENSP00000490105); hCLN3 isoform 2 (transcript variant 2)—1876 nt (GenBank NM_000086, UniProtKB/Swiss-Prot Q13286, CCDS 10632, Ensembl-Tr: ENST00000359984 (transcript: CLN3-205), protein—ENSP00000353073).
Figure 1.
The schematic structure of two different full-length (complete) CLN3 mRNA isoforms transcribed from varying promoters of the CLN3 gene of Homo sapiens. Only regions corresponding to distinguished (differing) transcription start points and 2–3 of the first exons (out of 15–17) of the hCLN3 gene are shown. hCLN3 isoform 1 (transcript variant 1)—3832 nt (GenBank NM_001042432, UniProtKB/Swiss-Prot Q13286, CCDS 10632, Ensembl-Tr: ENST00000636147 (transcript: CLN3-244), protein—ENSP00000490105); hCLN3 isoform 2 (transcript variant 2)—1876 nt (GenBank NM_000086, UniProtKB/Swiss-Prot Q13286, CCDS 10632, Ensembl-Tr: ENST00000359984 (transcript: CLN3-205), protein—ENSP00000353073).

Figure 2.
The CLN3 protein mediates anterograde and retrograde trafficking of Golgi network—plasma membrane, participates in the formation of endosomes, autophagosomes, and lysosomes.
Figure 2.
The CLN3 protein mediates anterograde and retrograde trafficking of Golgi network—plasma membrane, participates in the formation of endosomes, autophagosomes, and lysosomes.

Figure 3.
Processes activating in cln3− cells in presence of unregulated signaling cascades.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Possible molecular mechanisms of the JNCL (juvenile neuronal ceroid lipofuscinosis).
| Physiological Changes in cln3− Cells and/or the Cellular System Affected | Probable Cause of the Cell Death | References |
|---|---|---|
| Endosomal-lysosomal system, autophagy | Defects in traffic of proteins, cholesterol, sphingolipids, neurotransmitters. Accumulation of damaged organelles | [43,44,45,46,47,48,49,50,51,52,53] |
| Stress of endoplasmic reticulum (ER stress) | Accumulation of misfolded proteins and, as a consequence, apoptosis | [54,55] |
| Dysfunction of mitochondria | Deficiency in energy-rich phosphates ATP and ADP | [56,57,58,59,60,61,62] |
| Reactive oxygen species (ROS) | Damage of proteins, nucleic acids, lipids, membranes and organelles, as a consequence — apoptosis | [63,64] |
| pH homeostasis and osmoregulation | Сell destruction due to the inability to throw out excess of water | [65,66,67,68,69] |
| The hyperactivation of glial cells and astrocytes | Stimulation of inflammation | [70,71] |
| Upregulated activation of signal cascades | Excessive amount of some proteins leads to toxicity. Apoptosis | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shematorova, E.K.; Shpakovski, G.V. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. Int. J. Mol. Sci. 2020, 21, 8055. https://doi.org/10.3390/ijms21218055
AMA Style
Shematorova EK, Shpakovski GV. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. International Journal of Molecular Sciences. 2020; 21(21):8055. https://doi.org/10.3390/ijms21218055
Chicago/Turabian StyleShematorova, Elena K., and George V. Shpakovski. 2020. "Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease" International Journal of Molecular Sciences 21, no. 21: 8055. https://doi.org/10.3390/ijms21218055
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.