
Enantioselective Total Synthesis of (R,R)-Blumenol B and d9-(R,R)-Blumenol B
by
 ,
,
Shi Min Tan
1, 
Shaun W. P. Rees
1 ,
,
Rebecca E. Jelley
1,
Jin Wang
1,
Bruno Fedrizzi
1,2,* and
David Barker
1,2,3,*
1
School of Chemical Sciences, University of Auckland, 23 Symonds St., Auckland 1010, New Zealand
2
Centre for Green Chemical Science, School of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1010, New Zealand
3
MacDiarmid Institute for Advanced Materials and Nanotechnology, Victoria University of Wellington, Wellington 6012, New Zealand
*
Authors to whom correspondence should be addressed.
Molecules 2022, 27(21), 7294; https://doi.org/10.3390/molecules27217294
Submission received: 18 September 2022
/
Revised: 18 October 2022
/
Accepted: 21 October 2022
/
Published: 27 October 2022
(This article belongs to the Special Issue Chemical Synthesis of Natural Products)
Abstract
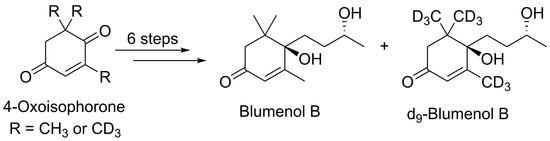
:C13-norisoprenoids are of particular importance to grapes and wines, as these molecules influence wine aroma and have been shown to significantly contribute to the distinct character of various wine varieties. Blumenol B is a putative precursor to a number of important wine aroma compounds, including the well-known compounds theaspirone and vitispirane. The enantioselective synthesis of (R,R)-blumenol B from commercially available 4-oxoisophorone was achieved using a short and easily scaleable route, which was then successfully applied to the synthesis of poly-deuterated d9-blumenol B.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Norisoprenoids are molecules found in grapes and wines that result from the direct degradation of carotenoids either photochemically, chemically or via oxidase-coupled mechanisms [1,2]. Those with 13 carbon atoms, i.e., C13-norisoprenoids, are of particular importance as these molecules, even at very low concentrations, are key contributors to the aroma profile of wine. C13-norisoprenoids are known to influence the distinctive sensory character of many wine varieties including Cabernet Sauvignon, Chenin blanc, Sauvignon blanc, Syrah and Riesling [3,4,5].
C13-norisoprenoids such as β-damascenone, α-ionol, β-ionol, α-ionone, β-ionone, vitispirane, actinidol, vomifoliol, and TDN (1,1,6-trimethyl-1,2-dihydronaphthalene) have been studied extensively not only in grapes and wines [6,7,8,9], but in other natural sources including honey, fruit and essential oils [1,10,11]; on the other hand, studies of blumenol B remain scarce.
Blumenol B 1 (also known as 7,8-dihydrovomifoliol), in its aglycone form, has been identified following the extraction and subsequent enzymatic hydrolysis of the non-volatile glycosylate precursor, icariside B5, which is present in the grapes and wines of Weisser Riesling [4], and also in Riesling grapevine leaves [3]. Blumenol B 1 has also been obtained via glycosylated precursor iscariside B5 from other natural sources, including Pinus sylvestris and Picea abies needles [12], Casearia sylvestris leaves [13], and Sarcandra glabra Nakai [14]. Furthermore, blumenol B 1 has been directly isolated from the leaves of Podocarpus blumei [15], and Cannabis sativa [16].
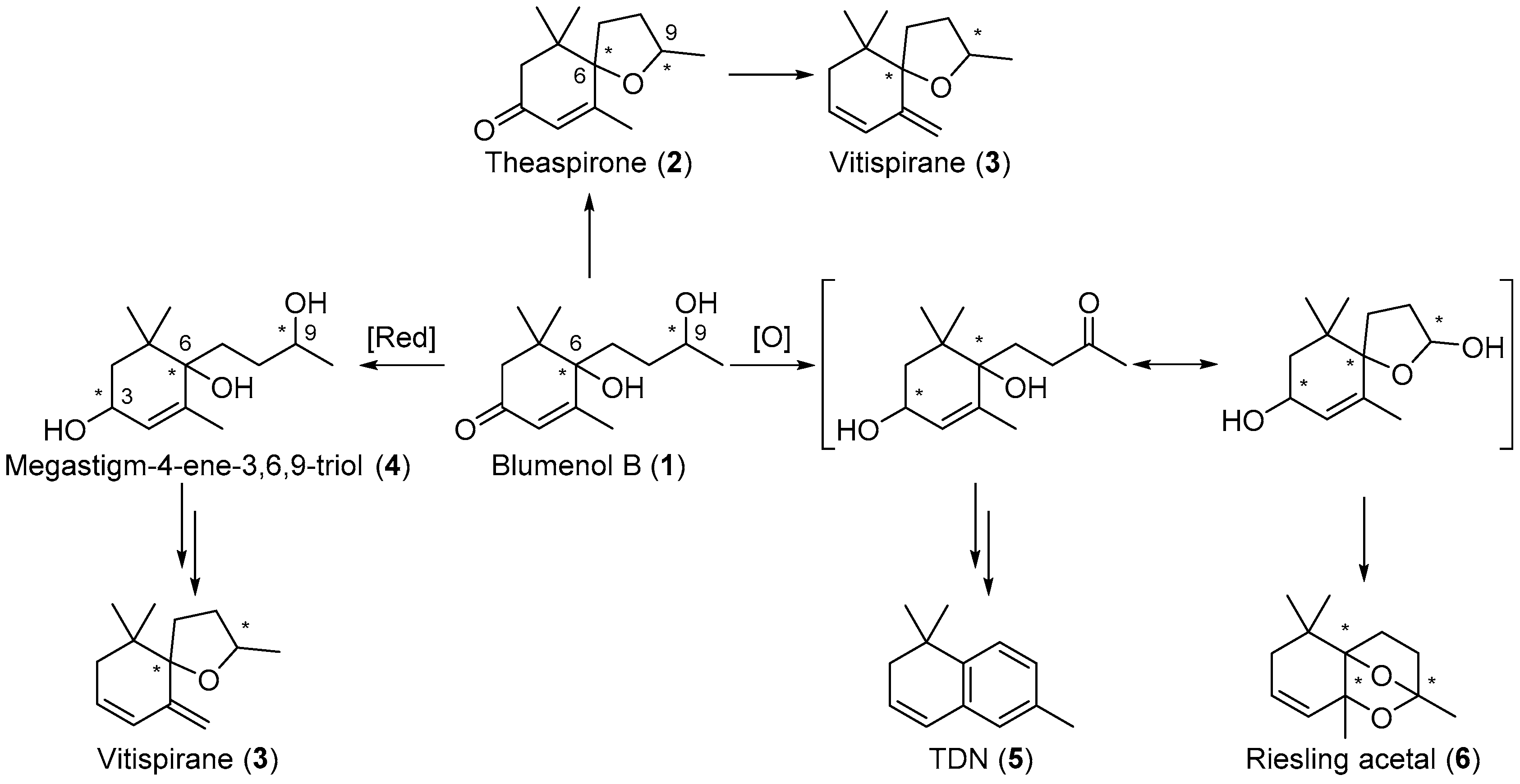
Blumenol B 1 is reported to be a precursor for other important aroma compounds and, in its reduced and oxidised forms, has been reported to prompt the formation of other C13-norisoprenoids, including theaspirone 2 [17], vitispirane 3 [18], megastigm-4-ene-3,6,9-triol 4, 1,1,6-trimethyl-1,2-dihydronaphthalene (TDN) 5 [19], and Riesling acetal 6 (Scheme 1) [20]. Stereochemistry is important in these aroma compounds as the various stereoisomers of these compounds contribute to the distinct aroma profiles of various grapes and wines [6,7,21,22,23,24]. For example, the (S,S)-stereoisomer of theaspirone 2 imparts an earthy scent, while the enantiomer gives off a sweet and tea-like odour [18,25,26]. The racemic mixture of vitispirane 3 is reported to have a woody, balsamic, resinous, and spicy aroma profile [18,27]. Pure isomers (R,R)- and (S,S)-vitispirane exert a floral and fruity aroma, while (S,R)- and (R,S)-vitispirane imparts a strong scent of exotic flowers and woody tone [6]. The aroma profile of another aroma compound, Riesling acetal 6, also differs between its enantiomeric forms, (+)-Riesling acetal imparts a subtle floral, fruity and woody scent, while (−)-Riesling acetal gives off a weak floral and camphoraceous aroma [1,19,28]. It is proposed that the stereochemistry of the precursor blumenol B 1 is important in the formation of the various isomers of 2, 3, and 6, and is thus important in the overall aroma profile of the wines.
To date, blumenol B 1 has only been synthesized via routes that were either non-stereospecific, lengthy (17-steps) or involved a semi-synthetic route from complex isolated natural products, such as the case of (S,R)- and (R,S)-blumenol B [17,25,29]. We wished to develop an efficient stereospecific synthesis of blumenol B from commercially available materials that would also be suitable for the preparation of isotopically labelled standards. Such standards could be employed for the identification and quantification of aglycones that are enzymatically and/or biochemically released from glycosidic precursors found in wine.
2. Results
A previous synthesis of structurally similar C13-norisoprenoids such as (±)-theaspirone and (±)-(Z)-vomifoliol utilized the coupling of a organometallic reagent with a cyclic ketone to form the key carbon–carbon bond at the tertiary alcohol center [17,25]. These previously endeavors were only able to form ring-opened derivatives such as 1 and 4 by an acidic ring cleavage of spiro-like molecules similar to 2. This not only resulted in ring-opened compounds but numerous rearranged and dehydrated species. We aimed to employ a similar organometallic strategy of addition to a ketone when attempting the racemic synthesis of blumenol B 1, but would avoid the formation of spiro-compounds enroute to 1. We began using the commercially available 4-oxoisophorone 7 as the substrate for coupling with a lithiated acetylide. The first step in this pathway was protection of the less hindered carbonyl in 4-oxoisophorone 7 to afford the known ketal 8, which was reacted with lithiated TMS-acetylene giving tertiary alcohol 9 in 65% yield across the two steps (Scheme 2) [30,31]. Removal of the TMS-group formed the free acetylide 10, which was reacted with acetaldehyde to afford diol 11 as an inseparable mixture of diastereomers. Acetylene 11 then underwent exhaustive hydrogenation of the exocyclic alkyne, followed by deprotection of the ketone acetal to give (±)-blumenol B 1 as an inseparable 2:1 diastereomeric mixture in 53% yield over two steps. It was found that hydrogenation for 20 h reaction time resulted in an exclusive reactive of the endocyclic alkene [32,33]. However, longer reaction times were found to result in unwanted over-reduction. The spectroscopic data of the mixture of isomers of (±)-1 were in agreement with those reported by Matsunami et al. [29] (Tables S1 and S2).
Following the development of this short racemic synthesis of 1, the next goal was the stereoselective synthesis of (R,R)-blumenol B using enantiopure reagents to direct the stereochemistry of the two chiral centers (Scheme 3). The first enantiopure reagent employed was commercially available (R)-(+)-3-butyn-2-ol 12, which was protected with a TBDPS group giving 13 in quantitative yield [34]. The second enantiopure reagent used was 2R,3R-(-)-2,3-butanediol, which was used to protect the less-hindered carbonyl on 4-oxoisophorone 7, forming the known chiral ketal 14 [35]. Alkynylation of ketal 14 using lithiated 13 afforded a 1.2:1 diastereomeric mixture of the R- and S- configuration at the newly formed tertiary alcohol stereocenter. Subsequent recrystallization using petroleum ether gave the major R-isomer 15 in 47% yield as a white solid and minor S-isomer 15a in 12% yield as a colourless oil, allowing for 15 to be easily separated. Similar facial selectivity, and solid versus oil for the two diastereoisomers, has been observed for the additional of alternative acetylenes with chiral ketone 14, which allowed for the determination of absolute stereochemistry in 15 [35]. This was further confirmed when crystals of d9-16 were formed (see below) and X-ray unequivocally determined the absolute stereochemistry. With 15 formed, both the acetal group and TBDPS-protecting group were removed to form propargyl alcohol 17 in 91% yield, across the two steps. The hydrogenation of propargyl alcohol 17 in the presence of Lindlar catalyst afforded (R,R)-blumenol B 1 in 57% yield, which exhibited identical 1H and 13C NMR data to reported values for (S,S)-blumenol B (Table S3) [29].
Following the successful synthesis of (R,R)-blumenol B 1, the next goal was to apply this methodology to synthesize isotopically labelled (R,R)-1, for possible use as an analytical standard for quantifying (R,R)-1. This would require the synthesis of an isotopically labelled precursor and it was decided to use the previously reported d9-8 in this study [36]. The synthesis began with treating 1,4-cyclohexanedione monoethylene acetal 18 with four equivalents of CD3I, which afforded the target tri-substituted d9-19 in 77% (Scheme 4). Next, the α,β-unsaturated ketone was installed in two steps by converting d9-19 to the TMS-protected enol d9-20, which was then subjected to one-pot halogenation and elimination, using Br2 followed by DBU to give the target enone d9-8 in 70% yield over the two steps. Subsequent removal of the acetal group using 2M HCl afforded d9-7, which could then be utilized for synthesis of d9-(R,R) blumenol B 1 via the aforementioned methods. In brief, d9-7 was protected using 2R,3R-(-)-2,3-butanediol to give chiral ketal d9-14, which was then reacted with acetylide 13 affording d9-15 in 44% yield over the two steps. Chiral acetal d9-15 was removed using 2M HCl and subsequent TBDPS deprotection with TBAF gave d9-17 in 94% yield. X-ray diffraction studies on TBDPS-protected d9-16 intermediate confirmed the desired R,R diastereomer had formed (Figure S1). Lastly, the alkyne motif was fully hydrogenated using H2 and Lindlar catalyst, forming d9-(R,R) blumenol B 1 in 44% yield.
3. Materials and Methods
3.1. General Experimental Details
All reactions in non-aqueous solvents were carried out under an inert atmosphere using anhydrous AR grade solvents. Solvents used for reaction work up and purification were used as purchased, without further purification. Thin-layer chromatography (TLC) was performed using Merck silica gel F354 aluminium plates pre-coated with silica. Flash chromatography was carried out using Silica Gel 60 (40–63 μm, 230–430 mesh ASTM) utilising solvent systems defined in the experimental procedure for each synthesized molecule. Infra-red (IR) spectra were obtained using a Perkin-Elmer Spectrum 1000 series Fourier Transform Infra-Red ATR spectrometer. Melting points were measured using a Reicher-Kofler block and are uncorrected. NMR spectra were obtained using a Bruker Avance DRX 400 MHz spectrometer at ambient temperature. Chemical shifts are reported relative to the residual solvent peak of either CDCl3 (δ 7.26 for 1H and δ 77.16 for 13C) or CD3OD (δ 3.31 for 1H and δ 49.00 for 13C). 1H NMR data are reported in the following sequence: position (δ), relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; m, multiplet; br s, broad singlet; br d, broad doublet), coupling constant (J, Hz), and proton assignment. 13C NMR data were reported in the following sequence: position (δ), multiplicity (d, doublet; q, quartet), coupling constant (J, Hz), and the carbon assignment. NMR assignments were made using a combination of 1H NMR, 13C NMR, HSQC and HMBC experiments. High-resolution mass spectroscopy (HRMS) was carried out using electrospray ionisation (ESI) on a MicroTOF-Q mass spectrometer.
3.2. Synthesis of Compounds
4,4-(Ethylenedioxy)-2,6,6-trimethylcyclohex-2-en-1-one 8: A mixture of 4-oxoisophorone 7 (3.0 g, 19.7 mmol), ethylene glycol (1.46 mL, 26.2 mmol), and toluene-p-sulfonic acid monohydrate (0.09 g, 0.473 mmol) in toluene (14.8 mL) were heated at reflux overnight with a Dean-Stark trap. The solution was left to cooled and quenched with sat. aq. NaHCO3 (15 mL) and was extracted with diethyl ether (3 × 15 mL). The combined organic extracts were washed with water and dried over anhydrous MgSO4. The solvent was removed in vacuo and the crude product was purified by flash chromatography (1:1 hexanes, ethyl acetate) to yield the title compound 8 (3.325 g, 86%) as a yellow oil. RF (3:1 petroleum ether, ethyl acetate) = 0.54. δH (400 MHz; CDCl3) 1.20 (6H, s, 6-(CH3)2), 1.79 (3H, d, J = 1.39 Hz, 2-CH3), 2.08 (2H, s, 5-H), 3.98–4.02 (4H, m, OCH2CH2O), 6.30 (1H, br s, 3-H). The spectroscopic data were in agreement with the literature values [31].
4,4-(Ethylenedioxy)-2,6,6-trimethyl-1-trimethylsilanyl-ethynylcyclohex-2-en-1-ol (±)-9: To a stirred solution of trimethylsilylacetylene (0.29 mL, 2.03 mmol) in THF (3 mL) under an nitrogen atmosphere at −78 °C was added nBuLi (1.6 M in hexanes, 0.22 mL, 2.03 mmol). The reaction mixture was stirred at −78 °C for 15 min and a solution of 8 (0.20 mg, 1.01 mmol) in THF (6 mL) was added dropwise to the previously prepared reaction mixture. The resultant mixture was then stirred under a nitrogen atmosphere for 1 h, and the reaction was quenched with sat. aq. NH4Cl (5 mL), and was extracted with diethyl ether (3 × 5 mL), before the combined organic extracts were washed with water (5 mL), brine (5 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (14:1 hexanes, ethyl acetate) to yield the title compound (±)-9 (0.22 g, 75%) as a yellow oil. RF (14:1, 9:1 hexanes, ethyl acetate) = 0.39. δH (400 MHz; CDCl3) 0.16 (9H, s, Si(CH3)3), 1.05 and 1.12 (each 3H, s, 6-(CH3)2), 1.89 (2H, s, 5-H), 1.91 (3H, s, 2-CH3), 3.90–3.95 (4H, m, 4H, m, OCH2CH2O), 5.36 (1H, br s, 3-H). The spectroscopic data were in agreement with the literature values [31].
4,4-(Ethylenedioxy)-2,6,6-trimethyl-1-ethynylcyclohex-2-en-1-ol (±)-10: To a stirred solution of (±)-9 (1.80 g, 6.12 mmol) in methanol (50 mL), potassium carbonate (2.54 g, 18.8 mmol) was added, and the reaction mixture was stirred at room temperature for 1 h. The precipitate was filtered, the solvent was removed in vacuo, and was extracted with diethyl ether (3 × 50 mL); then, the combined organic extracts were washed with water, dried over anhydrous MgSO4 and the solvent removed in vacuo. The crude product was recrystallised from hexanes to yield the title compound (±)-10 (1.85 g, 64%) as white needles. RF (9:1 hexanes, ethyl acetate) = 0.47. Melting point: 76–78 °C (lit. 78–80 °C). δH (400 MHz; CDCl3) 1.10 (3H, s, 6-CH3), 1.16 (3H, s, 6-CH3), 1.88 (1H, d, J = 14.3 Hz, 5-Ha), 1.93 (3H, d, J = 1.3 Hz, 2-CH3), 1.96 (1H, d, J = 14.2 Hz, 5-Hb), 2.02 (1H, s, OH), 2.50 (1H, s, ethynyl CH), 3.90–3.96 (4H, m, OCH2CH2O), 5.38 (1H, s, 3-H). The spectroscopic data and melting point were in agreement with the literature values [31].
8-(3-Hydroxybut-1-yn-1yl)-7,9,9-trimethyl-1,4-dioxaspiro [4.5]dec-6-en-8-ol (±)-11: To a stirred solution of (±)-10 (0.85g, 3.8 mmol) in THF (25 mL) under anitrogen atmosphere, LDA (5.71 mL, 11.4 mmol) was added at −78 °C, and the resultant mixture was stirred for 30 min. Acetaldehyde (0.327 mL, 5.85 mmol) in THF (12 mL) was added dropwise to the reaction mixture at −78 °C and was left to stir for 30 min; it was then further stirred at 0 °C under a nitrogen atmosphere overnight. The reaction mixture was quenched with sat. aq. NH4Cl (15 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine (25 mL), dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:1 hexanes, ethyl acetate) to yield the title compound (±)-10 (0.624 g, 62%) as a white solid. RF (1:1 hexanes, ethyl acetate) = 0.3. Melting point: 135–138 °C. δH (400 MHz; CDCl3) 1.09 (3H, s, 9-CH3), 1.14 (3H, s, 9-CH3), 1.45 (3H, d, J = 6.5 Hz, CH(OH)CH3), 1.91 (3H, s, 7-CH3), 2.03 (2H, br d, J = 7.6 Hz, 5CH2), 3.90–3.97 (4H, m, OCH2CH2O), 4.54–4.57 (1H, m, CH(OH)CH3), 5.36 (1H, br s, 3-H). The 1H NMR spectroscopic data and melting point were in agreement with the literature values [25].
(±)-Blumenol B (±)-1: To a stirred solution of (±)-11 (300 mg, 1.13 mmol) in MeOH (30 mL), Pa/BaSO4 (120 mg, 40% w/w) was added, and was stirred under a hydrogen atmosphere at room temperature for 18–20 h. The mixture was filtered through Celite®, washed with MeOH and the solvent was removed in vacuo to yield the alkane (160 mg, 53%) as a colorless oil, which was immediately dissolved in THF (8 mL). The solution was placed under a nitrogen atmosphere, 2 M HCl (0.8 mL) was added, and the resultant mixture was stirred at room temperature overnight. The solvent was removed in vacuo and extracted with ethyl acetate (3 × 5 mL), and the combined organic extracts were washed with brine (5 mL), and dried over anhydrous MgSO4, and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:3 hexanes, ethyl acetate) to yield the title compound (±)-1 (153 mg, 96%) as a colorless oil. RF (1:1 hexanes, ethyl acetate) = 0.15. δH (400 MHz; CD3OD) 1.02 (3H, s, 1-H), 1.10 (3H, s, 12-H), 1.22 (3H, d, J = 6.0 Hz, 10-H), 1.40–1.48 (1H, m, 8-H), 1.68–1.72 (1H, m, 8-H), 1.75–1.83 (1H, m, 7-H), 1.92–1.99 (1H, m, 7-H), 2.04 (3H, s, 13-H), 2.16 (1H, d, J = 18.1 Hz, 2-H), 2.59 (1H, dd, J = 4.5, 18.2 Hz, 2-H), 3.66 (1H, qui, J = 5.7 Hz, 9-H), 5.83 (1H, s, 4-H). δC (100 MHz; CD3OD) 21.8 (C-13), 23.7 (C-10), 24.0 (C-12), 24.5 (C-11), 35.3 (C-8), 35.7 (C-7), 43.0 (C-1), 51.1 (C-2), 69.3 (C-9), 79.2 (C-6), 126.6 (C-4), 171.8 (C-5), 200.8 (C-3). HRMS (ESI+): Found (MNa+): 249.1457 C13H22NaO3 requires 249.1461. IR: νmax(film)/cm−1; 3405 (O-H), 2965 (C-H), 2927 (C-H), 2878 (C-H), 2530 (O-H), 1647 (C=O), 1417 (C=C). Other diastereomer: δH (400 MHz; CD3OD) 1.02 (3H, s, 11-H), 1.10 (3H, s, 12-H), 1.22 (3H, d, J = 6.0 Hz, 10-H), 1.40–1.48 (1H, m, 8-H), 1.68–1.72 (1H, m, 8-H), 1.75–1.83 (1H, m, 7-H), 1.92–1.99 (1H, m, 7-H), 2.04 (3H, s, 13-H), 2.16 (1H, d, J = 18.1 Hz, 2-H), 2.59 (1H, dd, J = 4.5, 18.2 Hz, 2-H), 3.76–3.78 (1H, m, 9-H), 5.88 (1H, s, 4-H). δC (100 MHz; CD3OD) 21.7 (C-13), 23.5 (C-10), 24.0 (C-12), 24.6 (C-11), 35.2 (C-8), 35.6 (C-7), 42.9 (C-1), 51.1 (C-2), 68.9 (C-9), 79.1 (C-6), 126.6 (C-4), 171.7 (C-5), 200.9 (C-3). HRMS (ESI+): Found (MNa+): 249.1457 C13H22NaO3 requires 249.1461. IR: νmax(film)/cm−1; 3405 (O-H), 2965 (C-H), 2927 (C-H), 2878 (C-H), 2530 (O-H), 1647 (C=O), 1417 (C=C). The spectroscopic data were in agreement with the literature values [25,29].
(R)-(But-3-yn-2-yloxy)(tert-butyl)diphenylsilane (R)-13: To a stirred solution of (R)-(+)-3-butyn-2-ol (300 mg, 4.28 mmol) in DMF (15 mL) under an nitrogen atmosphere, imidazole (870 mg, 12.8 mmol) and TBDPSCl (1.66 mL, 6.40 mmol) was added at 0 °C, and was stirred for at room temperature for 4 h. The reaction mixture was quenched with sat. aq. NaHCO3 (7 mL), and was extracted with ethyl acetate (3 × 15 mL), the combined organic extracts were washed with water (3 × 15 mL), dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (9:1 hexanes, ethyl acetate) to yield the title compound (R)-13 (980 mg, 99%) as a colourless oil. RF (9:1 hexanes, ethyl acetate) = 0.72. δH (400 MHz; CDCl3) 1.09 (9H, s, Si(CH3)), 1.40 (3H, d, J = 6.5 Hz, 1-CH3), 2.34 (1H, d, J = 2.5 Hz, 4-H), 4.47 (1H, qd, J = 6.5, 2.0 Hz, 2-H), 7.36–7.46 (6H, m, Ar-H), 7.60–7.81 (4H, m, Ar-H). +84.4° (c = 1.00, MeOH). The spectroscopic data were in agreement with the literature values [34].
(2R,3R)-2,3,7,9,9-Pentamethyl-1,4-dioxaspiro [4.5]dec-6-en-8-one (R,R)-14: To a stirred solution of 4-oxoisophorone 7 (300 mg, 1.97 mmol) in toluene (8 mL), 2R,3R-(+)-2,3-butanediol (236 mg, 2.62 mmol) and p-toluenesulfonic acid (8.15 mg, 0.0473 mmol) was added. The reaction mixture was heated at reflux for 24 h with a Dean-Stark trap for the removal of excess water. The reaction mixture was cooled and quenched with sat. aq. NaHCO3 (8 mL), and then extracted with diethyl ether (3 × 8 mL), dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (14:1 petroleum ether, ethyl acetate) to yield the title compound (R,R)-14 (280 mg, 93%) as a pale yellow oil. RF (3:1 petroleum ether, ethyl acetate) = 0.75. δH (400 MHz; CDCl3) 1.17 (3H, s, 11-H), 1.22 (3H, s, 12-H), 1.27–1.30 (6H, m, 14 and 15-H), 1.79 (3H, d, J = 1.5 Hz, 13-H), 2.03–2.07 (1H, dd, J = 14.0, 1.5 Hz, 1H, 10-H), 2.10–2.13 (1H, d, J = 14.0 Hz, 10-H), 3.61–3.71 (2H, m, 2 and 3-H), 6.33 (1H, t, J = 1.3 Hz, 6-H). −16.1 (c = 0.97, MeOH). The spectroscopic data were in agreement with the literature values [35].
(2R,3R,8R)-8-((R)-3′((tert-Butyldiphenylsilyl)oxy)but-1′-yn-1′-yl-2,3,7,9,9-pentamethyl-1,4-dioxaspiro [4.5]dec-6-en-8-ol (R,R)-15: To a stirred solution of (R)-13 (288 mg, 0.89 mmol) in THF (8 mL) under an nitrogen atmosphere, and nBuLi (2 M in hexanes, 0.45 mL, 0.89 mmol) was added at −78 °C. The resultant mixture was stirred at −78 °C for 20 min and a solution of (R,R)-14 (100 mg, 0.45 mmol) in THF (4 mL) was added dropwise. The mixture was further stirred under a nitrogen atmosphere at room temperature overnight. The reaction was quenched with sat. aq. NH4Cl (6 mL) and was extracted with diethyl ether (3 × 10 mL), the combined organic extracts were washed with water (6 mL), brine (6 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was recrystallised from petroleum ether to yield the title compound (R,R)-15 (112 mg, 47%) as a white solid. Melting point: 95–99 °C. δH (400 MHz; CDCl3) 1.02 (3H, s, 11-H), 1.06 (12H, br s, 12-H and SiC(CH3)), 1.22 (6H, t, J = 5.2 Hz, 16 and 17-H), 1.41 (3H, d, J = 6.5 Hz, 10-H), 1.70 (3H, d, J = 1.35 Hz, 13-H), 1.76 (1H, d, J = 14.1, 2-H), 1.93 (1H, d, J = 14.1, 2-H), 1.86 (2H, dd, J = 59.6, 14.2 Hz, 2-H), 3.50–3.60 (2H, m, 14 and 15-H), 4.52 (1H, q, J = 6.5 Hz, 9-H), 5.30 (1H, s, 4-H), 7.36–7.46 (6H, m, Ar-H), 7.70 (2H, dd, J = 8.0, 2.0 Hz, Ar-H), 7.76 (2H, dd, J = 8.0, 2.0 Hz, Ar-H). δC (100 MHz; CDCl3) 14.3 (C-13), 18.9 (C-16 and 17), 22.3 (C-11 and 12), 25.5 (C-10), 26.9 (SiC(Ph)2C(CH3)3), 39.4 (SiC(Ph)2C(CH3)3), 45.6 (C-2), 60.1 (C-9), 74.3 (C-7), 78.0 (C-14 and 15), 84.1 (C-8), 88.1 (C-6), 104.1 (C-3), 124.8 (C-4), 136.0 (Si(CH3)), 140.4 (C-5). HRMS (ESI+): Found (MNa+): 555.2898 C33H44NaO4Si requires 555.2901.
(R)-6-Hydroxy-6-((R)-9-hydroxybut-7-yn-7-yl)-1,1,5-trimethylcyclohex-4-en-3-one (R,R)-17: To a stirred solution of (R,R)-15 (703 mg, 1.32 mmol) in THF (40 mL) under a nitrogen atmosphere, 2M HCl (6 mL) was added and the mixture stirred under an nitrogen atmosphere at room temperature overnight. The solvent was removed in vacuo and the remaining residue was extracted with ethyl acetate (3 × 40 mL). The combined organic extracts were washed brine (40 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. This crude intermediate (R,R)-16 was dissolved in THF (40 mL) under a nitrogen atmosphere, TBAF (0.73 mL, 2.52 mmol) was added at 0 °C. The resultant mixture was stirred under a nitrogen atmosphere at room temperature overnight. The reaction was quenched with water (40 mL), and the solvent was removed in vacuo, and was extracted with ethyl acetate (3 × 40 mL), the combined organic extracts were washed brine (40 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:3 petroleum ether, ethyl acetate) to yield the title compound (R,R)-17 (270 mg, 91%) as a colourless oil. N.B. compound 17 was found to be very volatile and prone to evaporation if placed under strong vacuum. RF (1:1 petroleum ether, ethyl acetate) = 0.23. δH (400 MHz; CDCl3) 1.09 (3H, s, 11-H), 1.19 (3H, s, 12-H), 1.47 (3H, d, J = 6.5 Hz, 10-H), 2.10 (3H, d, J = 1.35 Hz, 13-H), 2.38 (1H, d, J = 16.5 Hz, 2-H), 2.51 (1H, d, J = 16.5 Hz, 2-H), 4.58 (1H, q, J = 6.6, 9-H), 5.83 (1H, s, 4-H). δC (100 MHz; CDCl3) 19.8 (C-12), 22.8 (C-11), 24.4 (C-10), 25.2 (C-13), 29.7 (C-1), 49.4 (C-2), 58.4 (C-9), 74.5 (C-7), 82.8 (C-5), 89.4 (C-8), 126.3 (C-4), 160.4 (C-5), 198.4 (C-3). HRMS (ESI+): Found (MNa+): 245.2712 C13H18NaO3 requires 245.2712. = −138.79° (c = 0.66, MeOH). IR: νmax(film)/cm−1; 3456, 3025, 2864, 1396, 1086.
(6R,9R)-Blumenol B (R,R)-1: To a stirred solution of (R,R)-17 (14 mg, 0.0629 mmol) in MeOH (5 mL), Pd/BaSO4 (1.4 mg, 20% w/w) was added, and the resultant mixture was stirred under a hydrogen atmosphere overnight. The mixture was filtered through Celite®, washed with MeOH and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:2 petroleum ether, ethyl acetate) to yield the title compound (R,R)-1 (8 mg, 57%) as a colourless oil. RF (1:3 petroleum ether, ethyl acetate) = 0.26. δH (400 MHz; CD3OD) 1.02 (3H, s, 11-H), 1.09 (3H, s, 12-H), 1.16 (3H, d, J = 6.2 Hz, 10-H), 1.40 (1H, dddd, J = 12.7, 11.5, 8.05, 5.12 Hz, 8-H), 1.68 (1H, tt, J = 12.7, 4.5 Hz, 8-H), 1.79 (1H, td, J = 12.7, 4.0 Hz, 7-H), 1.98 (1H, td, J = 12.7, 5.3 Hz, 7-H), 2.04 (3H, d, J = 1.49 Hz, 13-H), 2.16 (1H, dd, J = 18.0, 1.02 Hz, 2-H), 2.59 (1H, d, J = 18.1 Hz, 2-H), 3.61–3.69 (1H, m, 9-H), 5.83 (1H, s, 4-H). δC (100 MHz; CD3OD) 21.8 (C-13), 23.7 (C-10), 24.0 (C-11), 24.5 (C-12), 35.3 (C-7), 35.7 (C-8), 42.9 (C-1), 51.5 (C-2), 69.3 (C-9), 79.2 (C-6), 126.6 (C-4), 171.8 (C-5), 200.8 (C-3). HRMS (ESI+): Found (MNa+): 249.3115 C13H22NaO3 requires 249.3115. = −1.25° (c = 0.16, MeOH). IR: νmax(film)/cm−1; 3469, 3068, 2956, 1410, 1097.
Compounds d9-19, d9-20 and d9-8 were prepared using the reported method [36].
2,6,6-Tris(methyl-d3)cyclohex-2-ene-1,4-dione-d9 d9-7: To a stirred solution of d9-8 (61 mg, 0.297 mmol) in THF (7 mL) under a nitrogen atmosphere, 2M HCl (3.5 mL) was added, and the resultant mixture was stirred under a nitrogen atmosphere at room temperature overnight. The solvent was removed in vacuo and was extracted with ethyl acetate (3 × 7 mL) and the combined organic extracts were washed with brine (7 mL), and dried over anhydrous MgSO4, and the solvent was removed in vacuo. The crude product was purified by flash chromatography (4:1 petroleum ether, ethyl acetate) to yield the title compound d9-7 (48 mg, quant.) as a colourless oil. RF (3:1 petroleum ether, ethyl acetate) = 0.61. δH (400 MHz; CDCl3) 2.70 (2H, s, 5-H), 6.55 (1H, s, 3-H). δC (100 MHz; CDCl3) 44.9 (C-6), 51.8 (C-5), 137.2 (C-3), 149.0 (C-2), 197.9 (C-4), 203.7 (C-1). HRMS (ESI+): Found (MNa+): 184.2413 C9H3D9NaO2 requires 184.2412. IR: νmax(film)/cm−1; 3049, 2986, 2912, 1768, 1087.
(2R,3R)-2,3-dimethyl-7,9,9-tris(methyl-d3)-1,4-dioxaspiro [4.5]dec-6-en-8-one-d9 d9-(R,R)-14: To a stirred solution of d9-7 (1 g, 6.20 mmol) in toluene (15 mL), a solution of 2S,3S-(+)-2,3-butanediol (0.68 mL, 7.44 mmol) and p-toluenesulfonic acid (37 mg, 0.217 mmol) was added, and the resultant mixture was heated at reflux for 24 h with a Dean–Stark trap for the removal of excess water. The reaction mixture was cooled and quenched with sat. aq. NaHCO3 (15 mL), and was extracted with diethyl ether (3 × 8 mL), dried over anhydrous MgSO4 and the solvent removed in vacuo. The crude product was purified by flash chromatography (14:1 petroleum ether, ethyl acetate) to yield the title compound d9-(R,R)-14 (1.24 g, 89%) as a white solid. RF (9:1 petroleum ether, ethyl acetate) = 0.5. Melting point: 70–76 °C. δH (400 MHz; CDCl3) 1.28 (6H, t, J = 5.45 Hz, 11 and 12-H), 2.02–2.13 (2H, m, 10-H), 3.61–3.71 (2H, m, 2 and 3-H), 6.32 (1H, s, 6-H). δC (100 MHz; CDCl3) 16.7 and 16.9 (C-11 and 12), 47.5 (C-10), 78.5 (C-2 and 3), 103.0 (C-5), 141.5 (C-6), 204.6 (C-8). HRMS (ESI+): Found (MNa+): 256.3423 C13H11D9NaO3 requires 256.3428. + 5.50 (c = 0.4, CHCl3). IR: νmax(film)/cm−1; 3027, 2971, 1695, 1521, 1063.
(6R,14R,15R)-6-((R)-9-((tert-Butyldiphenylsilyl)oxy)but-7-yn-7-yl)-14,15-dimethyl-1,1,5-tris(methyl-d3)-18,19-dioxaspiro [3.19]dec-4-en-6-ol-d9 d9-(R,R)-15: To a stirred solution of (R)-13 (1.62 g, 5.04 mmol) in THF (30 mL) under a nitrogen atmosphere, nBuLi (2 M in hexanes, 2.52 mL, 5.04 mmol) was added at −78 °C. The reaction mixture was further stirred at −78 °C for 20 min and d9-(R,R)-14 (588 mg, 2.52 mmol) in THF (30 mL) was added dropwise. The resultant mixture was then further stirred at room temperature under a nitrogen atmosphere overnight. The reaction was quenched with sat. aq. NH4Cl (30 mL) and was extracted with ethyl acetate (3 × 30 mL), the combined organic extracts were washed with water (30 mL), brine (30 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (9:1 petroleum ether, ethyl acetate) and recrystallised from petroleum ether to yield the title compound d9-(R,R)-15 (1.98 g, 51%) as a white solid. RF (4:1 petroleum ether, ethyl acetate) = 0.41. Melting point: 111–115 °C. δH (400 MHz; CDCl3) 1.05 (9H, s, Si(CH3)), 1.20–1.23 (6H, t, J = 4.8 Hz, 16 and 17-H), 1.41 (3H, d, J = 6.5 Hz, 10-H), 1.52 (1H, br s, OH), 1.74 (1H, d, J = 14.2 Hz, 2-H), 1.92 (1H, d, J = 14.1 Hz, 2-H), 3.50–3.60 (2H, m, 14 and 15-H), 4.52 (1H, q, J = 6.4 Hz, 9-H), 5.28 (1H, s, 4-H), 7.34–7.44 (6H, m, Ar-H), 7.68–7.75 (4H, m, Ar-H). δC (100 MHz; CDCl3) 16.8 (C-16), 16.9 (C-17), 19.2 (2 x CD3), 25.3 (C-10), 26.7 ((-OSi(Ph-C)C(CH3)3)), 26.9 and 39.0 (-OSi(Ph-C)C(CH3)3)), 45.5 (C-2), 60.1 (C-9), 74.3 (C-7), 77.9 and 78.0 (C-14 and 15), 84.1 (C-8), 88.1 (C-6), 104.1 (C-3), 124.8 (C-4), 127.6–136.0 (-OSi(Ph-C)C(CH3)3)), 140.3 (C-5). HRMS (ESI+): Found (MNa+): 564.8435 C33H35D9NaO4Si requires 564.8435. = + 2.27° (c = 0.44, CHCl3). IR: νmax (film)/cm−1; 3574, 3058, 2871, 2133, 1739, 1524, 1088.
(R)-6-((R))-9-((tert-Butyldiphenylsilyl)oxy)but-7-yn-7-yl)-6-hydroxy-1,1,5-tris(methyl-d3)cyclohex-4-en-3-one-d9 d9-(R,R)-16: To a stirred solution of d9-(R,R)-15 (1.98 g, 3.71 mmol) in THF (40 mL) under a nitrogen atmosphere, 2M HCl (10 mL) was added, and the reaction mixture was stirred under anitrogen atmosphere at room temperature overnight. The solvent was removed in vacuo and was extracted with ethyl acetate (3 × 40 mL); the combined organic extracts were washed with brine (40 mL), dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (3:1 petroleum ether, ethyl acetate) to yield the title compound d9-(R,R)-16 (1.60 g, 94%) as white crystals. RF (3:1 petroleum ether, ethyl acetate) = 0.51. Melting point: 105–107 °C. δH (400 MHz; CDCl3) 1.05 (9H, s, Si(CH3)), 1.46 (3H, d, J = 6.5 Hz, 10-H), 2.25 (1H, d, J = 16.5 Hz, 2-H), 2.34 (1H, d, J = 16.5 Hz, 2-H), 4.57 (1H, q, J = 6.5 Hz, 9-H), 5.73 (1H, s, 4-H), 7.36–7.46 (6H, m, Ar-H), 7.70 (4H, dt, J = 20.4, 6.1, 1.5 Hz, Ar-H). δC (100 MHz; CDCl3) 19.2 (C-1), 25.2 (C-10), 26.8 ((-OSi(Ph-C)C(CH3)3)), 41.2 (C-2), 59.9 (C-9), 74.2 (C-6), 82.4 (C-8), 89.7 (C-7), 126.1 (C-4), 127.7 and 127.9 (-OSi(Ph-C)C(CH3)3)), 130.0 (-OSi(Ph-C)C(CH3)3)), 135.8 and 136.0 (-OSi(Ph-C)C(CH3)3)), 160.0 (C-5), 198.3 (C-3). HRMS (ESI+): Found (MNa+): 492.2878 C29H27D9NaO3Si requires 492.2891. = −2.35° (c = 0.34, CHCl3). IR: νmax(film)/cm−1; 3369, 3056, 2863, 1786, 1121.
(R)-4-Hydroxy-4-((R)-3′-hydroxybut-1-yn-1-yl)-3,5,5-tris(methyl-d3)cyclohex-2-en-1-one-d9 d9-(R,R)-17: To a stirred solution of d9-(R,R)-16 (69 mg, 0.15 mmol) in THF (7 mL) under a nitrogen atmosphere, TBAF (0.09 mL, 0.30 mmol) was added at 0 °C, and the resultant mixture was stirred at room temperature under a nitrogen atmosphere overnight. The reaction was quenched with water (7 mL), the solvent was removed in vacuo and extracted with ethyl acetate (3 × 7 mL), the combined organic extracts were washed with brine (7 mL) and dried over anhydrous MgSO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:2 petroleum ether, ethyl acetate) to yield the title compound d9-(R,R)-17 (35 mg, quant.) as a colourless oil. N.B. compound 17 was found to be very volatile and was prone to evaporation if placed under a strong vacuum. RF (1:1 petroleum ether, ethyl acetate) = 0.26. δH (400 MHz; CDCl3) 1.46 (3H, d, J = 6.5 Hz, 10-H), 2.37 (1H, d, J = 16.4 Hz, 2-H), 2.49 (1H, d, J = 16.4 Hz, 2-H), 2.83 (OH), 4.58 (1H, q, J = 6.6 Hz, 9-H), 5.83 (1H, s, 4-H). δC (100 MHz; CDCl3) 24.4 (C-10), 49.2 (C-2), 58.4 (C-9), 74.4 (C-6), 82.7 (C-7), 89.3 (C-8), 126.2 (C-4), 198.5 (C-3). HRMS (ESI+): Found (MNa+): 254.1704 C13H9D9NaO3 requires 254.1713. = −150.51° (c = 0.78, MeOH). IR: νmax(film)/cm−1; 3594, 2861, 2236, 1811, 1067.
(6R,9R)-Blumenol B-d9 d9-(R,R)-1: To a stirred solution of d9-(R,R)-17 (54 mg, 0.233 mmol) in MeOH (7 mL), Pd/BaSO4 (10 mg, 20% w/w) was added, and was stirred under a hydrogen atmosphere for 6 h. The mixture was filtered through Celite®, washed with MeOH and the solvent was removed in vacuo. The crude product was purified by flash chromatography (1:3 petroleum ether, ethyl acetate) to yield the title compound d9-(R,R)-1 as (24 mg, 44%) as a colourless oil. RF (1:3 petroleum ether, ethyl acetate) = 0.24. δH (400 MHz; CD3OD) 1.17 (3H, d, J = 6.2 Hz, 10-H), 1.34–1.44 (1H, m, 8-H), 1.64–1.82 (2H, m, 8-H), 1.94–2.02 (1H, m, 7-H), 2.15 (1H, dd, J = 18.1, 0.98 Hz, 2-H), 2.59 (1H, d, J = 18.1 Hz, 2-H), 3.62–3.69 (1H, m, 9-H), 5.83 (1H, s, 4-H). δC (100 MHz; CD3OD) 23.7 (C-10), 35.3 (C-7), 35.7 (C-8), 50.9 (C-2), 69.4 (C-9), 79.2 (C-6), 126.6 (C-4), 171.7 (C-5), 200.9 (C-3). HRMS (ESI+): Found (MNa+): 258.2018 C13H13D9NaO3 requires 258.2026. = −2.272° (c = 0.22, CHCl3). IR: νmax(film)/cm−1; 3412.1 (broad OH), 2850.9, 2917.2 and 2961.0 (C-H), 1650.9 and 1729.3 (C=O), 1275.4 (C-O).
4. Conclusions
In summary, we developed a six-step enantioselective route for the synthesis of (R,R) blumenol B 1. The synthesis is high-yielding, efficient, and significantly shorter than previously reported methods. Furthermore the method can be applied to the synthesis of d9-labelled (R,R)-blumenol B d9-(R,R)-1. The enantioselective synthesis involving the use of two readily available chiral reagents allowed for the selective formation and easy separation of respective stereoisomers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27217294/s1.
Author Contributions
Conceptualization, D.B. and B.F.; methodology, D.B., S.M.T., J.W.; investigation, S.M.T. and S.W.P.R.; resources, D.B., B.F.; writing—original draft preparation, S.M.T., S.W.P.R., J.W., D.B.; writing—review and editing, S.W.P.R., R.E.J., D.B., B.F.; supervision, D.B., B.F.; project administration, B.F. and D.B.; funding acquisition, D.B. and B.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Acknowledgments
We thank the University of Auckland for supporting this research.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Winterhalter, P.; Rouseff, R. Carotenoid-Derived Aroma Compounds: An Introduction. In Carotenoid-Derived Aroma Compounds; Winterhalter, P., Rouseff, R.L., Eds.; American Chemical Society: Washington, DC, USA, 2001; Volume 802, pp. 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hjelmeland, A.K.; Ebeler, S.E. Glycosidically Bound Volatile Aroma Compounds in Grapes and Wine: A Review. Am. J. Enol. Vitic. 2015, 66, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Skouroumounis, G.K.; Winterhalter, P. Glycosidically Bound Norisoprenoids from Vitis Vinifera Cv. Riesling Leaves. J. Agric. Food Chem. 1994, 42, 1068–1072. [Google Scholar] [CrossRef]
- Marais, J.; van Wyk, C.J.; Rapp, A. Effect of Sunlight and Shade on N Orisoprenoid Levels in Maturing Weisser Riesling and Chenin Blanc Grapes and Weisser Riesling Wines. South Afr. J. Enol. Vitic. 1992, 13, 23–32. [Google Scholar] [CrossRef]
- Ebeler, S.E.; Thorngate, J.H. Wine Chemistry and Flavor: Looking into the Crystal Glass. J. Agric. Food Chem. 2009, 57, 8098–8108. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pinto, M.M. Carotenoid Breakdown Products the—Norisoprenoids—In Wine Aroma. Arch. Biochem. Biophys. 2009, 483, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Winterhalter, P.; Schreier, P. C13-Norisoprenoid Glycosides in Plant Tissues: An Overview on Their Occurrence, Composition and Role as Flavour Precursors. Flavour Fragr. J. 1994, 9, 281–287. [Google Scholar] [CrossRef]
- Gök, R.; Bechtloff, P.; Ziegler, M.; Schmarr, H.-G.; Fischer, U.; Winterhalter, P. Synthesis of Deuterium-Labeled 1,1,6-Trimethyl-1,2-Dihydronaphthalene (TDN) and Quantitative Determination of TDN and Isomeric Vitispiranes in Riesling Wines by a Stable-Isotope-Dilution Assay. J. Agric. Food Chem. 2019, 67, 6414–6422. [Google Scholar] [CrossRef] [PubMed]
- Yamano, Y.; Ito, M. Synthesis of Optically Active Vomifoliol and Roseoside Stereoisomers. Chem. Pharm. Bull. 2005, 53, 541–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterhalter, P.; Schreier, P. Free and Bound C13 Norisoprenoids in Quince (Cydonia Oblonga, Mill.) Fruit. J. Agric. Food Chem. 1988, 36, 1251–1256. [Google Scholar] [CrossRef]
- Jerković, I.; Marek Kuś, P. Terpenes in Honey: Occurrence, Origin and Their Role as Chemical Biomarkers. RSC Adv. 2014, 4, 31710–31728. [Google Scholar] [CrossRef]
- Andersson, R.; Lundgren, L.N. Monoaryl and Cyclohexenone Glycosides from Needles of Pinus Sylvestris. Phytochemistry 1988, 27, 559–562. [Google Scholar] [CrossRef]
- Wang, W.; Li, X.-C.; Ali, Z.; Khan, I.A. Two New C13 Nor-Isoprenoids from the Leaves of Casearia Sylvestris. Chem. Pharm. Bull. 2009, 57, 636–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H. Isolation and Chemotaxonomic Significance of Megastigmane-Type Sesquiterpenoids from Sarcandra Glabra. J. Med. Plants Res. 2012, 6, 4501–4504. [Google Scholar] [CrossRef]
- Galbraith, M.N.; Horn, D.H.S. Structures of the Natural Products Blumenols A, B, and C. J. Chem. Soc. Chem. Commun. 1972, 113–114. [Google Scholar] [CrossRef]
- Ludwig Bercht, C.A.; Samrah, H.M.; Lousberg, R.J.J.C.; Theuns, H.; Salemink, C.A. Isolation of Vomifoliol and Dihydrovomifoliol from Cannabis. Phytochemistry 1976, 15, 830–831. [Google Scholar] [CrossRef]
- Marx, J.N. A New Synthesis of Theaspirone, an Odiferous Principle of Tea. Tetrahedron 1975, 31, 1251–1253. [Google Scholar] [CrossRef]
- Herion, P.; Full, G.; Winterhalter, P.; Schreier, P.; Bicchi, C. Enantiodifferentiation of Isomeric Vitispiranes. Phytochem. Anal. 1993, 4, 235–239. [Google Scholar] [CrossRef]
- Winterhalter, P. 1,1,6-Trimethyl-1,2-Dihydronaphthalene (TDN) Formation in Wine. 1. Studies on the Hydrolysis of 2,6,10,10-Tetramethyl-1-Oxaspiro[4.5]Dec-6-Ene-2,8-Diol Rationalizing the Origin of TDN and Related C13 Norisoprenoids in Riesling Wine. J. Agric. Food Chem. 1991, 39, 1825–1829. [Google Scholar] [CrossRef]
- Daniel, M.A.; Capone, D.L.; Sefton, M.A.; Elsey, G.M. Riesling Acetal Is a Precursor to 1,1,6-Trimethyl-1,2-Dihydronaphthalene (TDN) in Wine. Aust. J. Grape Wine Res. 2009, 15, 93–96. [Google Scholar] [CrossRef]
- Strauss, C.R.; Dimitriadis, E.; Wilson, B.; Williams, P.J. Studies on the Hydrolysis of Two Megastigma-3,6,9-Triols Rationalizing the Origins of Some Volatile C13 Norisoprenoids of Vitis Vinifera Grapes. J. Agric. Food Chem. 1986, 34, 145–149. [Google Scholar] [CrossRef]
- Dollmann, B.; Full, G.; Schreier, P.; Winterhalter, P.; Güntert, M.; Sommer, H. Synthesis and Enantiodifferentiation of Riesling Acetals. Phytochem. Anal. 1995, 6, 106–111. [Google Scholar] [CrossRef]
- Simpson, R.F.; Miller, G.C. Aroma Composition of Aged Riesling Wine. Vitis 1983, 22, 51–63. [Google Scholar]
- Simpson, R.F. 1,1,6-Trimethyl-1 2-Dihydronaphthalene: An Important Contributor to the Bottle Aged Bouquet of Wine. Chem. Ind. 1978, 1, 37. [Google Scholar]
- Weyerstahl, P.; Meisel, T. Structure-Odor Correlation, XIX. Synthesis and Olfactory Properties of Various Racemic Theaspirones, Ketoedulans and Edulans. Liebigs Ann. Chem. 1994, 1994, 415–427. [Google Scholar] [CrossRef]
- Nakatani, Y.; Yamanishi, T. Synthese Totale Des Cis-et Trans-1-Oxa-8-Oxo-2,6,10,10-Tetramethyl-Spiro(4:5)-6-Decenes. Tetrahedron Lett. 1969, 10, 1995–1998. [Google Scholar] [CrossRef]
- Schneider, R.; Razungles, A.; Augier, C.; Baumes, R. Monoterpenic and Norisoprenoidic Glycoconjugates of Vitis Vinifera L. Cv. Melon B. as Precursors of Odorants in Muscadet Wines. J. Chromatogr. A 2001, 936, 145–157. [Google Scholar] [CrossRef]
- Winterhalter, P.; Sefton, M.; Williams, P. New 13C-Norisoprenoid Intramolecular Acetal in Riesling Wine. Chem. Ind. Lond. 1990, 54, 463–464. [Google Scholar]
- Matsunami, K.; Otsuka, H.; Takeda, Y.; Miyase, T. Reinvestigation of the Absolute Stereochemistry of Megastigmane Glucoside, Icariside B5. Chem. Pharm. Bull. 2010, 58, 1399–1402. [Google Scholar] [CrossRef] [Green Version]
- Marx, J.N.; Sondheimer, F. Synthesis of (±)-Loliolide [(±)-Digiprolactone]. Tetrahedron 1966, 22, 1–7. [Google Scholar] [CrossRef]
- Hanson, J.R.; Uyanik, C. An Efficient Synthesis of the Plant Hormone Abscisic Acid. J. Chem. Res. 2003, 2003, 426–427. [Google Scholar] [CrossRef]
- Bogliotti, N.; Dalko, P.I.; Cossy, J. A One-Pot Process for the Enantioselective Preparation of Saturated Secondary Alcohols from Propargyl Ketones under Hydrogen Transfer Conditions. Tetrahedron Lett. 2005, 46, 6915–6918. [Google Scholar] [CrossRef]
- Hamed, O.; Henry, P.M.; Becker, D.P. Palladium(II)-Catalyzed Dicarboxymethylation of Chiral Allylic Alcohols: Chirality Transfer Affording Optically Active Diesters Containing Three Contiguous Chiral Centers. Tetrahedron Lett. 2010, 51, 3514–3517. [Google Scholar] [CrossRef]
- Kiyotsuka, Y.; Kobayashi, Y. Formation of Chiral C(Sp3)−C(Sp) Bond by Allylic Substitution of Secondary Allylic Picolinates and Alkynyl Copper Reagents. J. Org. Chem. 2009, 74, 7489–7495. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.R.; Clark, A.J.; Clarkson, G.J.; Taylor, P.C.; Marsh, A. Concise Enantioselective Synthesis of Abscisic Acid and a New Analogue. Org. Biomol. Chem. 2006, 4, 4186–4192. [Google Scholar] [CrossRef] [PubMed]
- Lamb, N.; Wahab, N.; Rose, P.A.; Shaw, A.C.; Abrams, S.R.; Cutler, A.J.; Smith, P.J.; Gusta, L.V.; Ewan, B. Synthesis, Metabolism and Biological Activity of a Deuterated Analogue of the Plant Hormone S-(+)-Abscisic Acid. Phytochemistry 1996, 41, 23–28. [Google Scholar] [CrossRef]
Scheme 1.
Blumenol B (1) is a precursor in the formation of theaspirone (2) and other important C13-norisoprenoids including vitispirane (3), megastigm-4-ene-3,6,9-triol (4), TDN (5) and Riesling acetal (6) in grapes and wines.
Scheme 1.
Blumenol B (1) is a precursor in the formation of theaspirone (2) and other important C13-norisoprenoids including vitispirane (3), megastigm-4-ene-3,6,9-triol (4), TDN (5) and Riesling acetal (6) in grapes and wines.

Scheme 2.
Synthesis of isomeric mixture of (±)-blumenol B 1. Reagents and conditions: (a) Ethylene glycol, p-TsOH, toluene, 24 h, 86%; (b) Trimethylsilylacetylene, nBuLi, THF, −78 °C, 16 h, 75%; (c) K2CO3, MeOH, 1 h, 64%; (d) Acetaldehyde, LDA, THF, −78 °C, 14 h, 62%; (e) Lindlar catalyst, H2, MeOH, 20 h then 2 M HCl, THF, 18 h, 52%.
Scheme 2.
Synthesis of isomeric mixture of (±)-blumenol B 1. Reagents and conditions: (a) Ethylene glycol, p-TsOH, toluene, 24 h, 86%; (b) Trimethylsilylacetylene, nBuLi, THF, −78 °C, 16 h, 75%; (c) K2CO3, MeOH, 1 h, 64%; (d) Acetaldehyde, LDA, THF, −78 °C, 14 h, 62%; (e) Lindlar catalyst, H2, MeOH, 20 h then 2 M HCl, THF, 18 h, 52%.

Scheme 3.
Synthesis of (R,R) blumenol B 1. Reagents and conditions; (a) TBDPS-Cl, imidazole, DMF, 0 °C, overnight, quant.; (b) 2R,3R-(-)-2,3-butanediol, p-TsOH, toluene, overnight, 93%; (c) nBuLi, THF, −78 °C, 14 h, 47% 15 and 12% 15a; (d) 2 M HCl, THF, 18 h, 95%; (e) TBAF, THF, 0 °C, 15 h, 96%; (f) Pd/BaSO4, H2, MeOH, 19 h, 57%.
Scheme 3.
Synthesis of (R,R) blumenol B 1. Reagents and conditions; (a) TBDPS-Cl, imidazole, DMF, 0 °C, overnight, quant.; (b) 2R,3R-(-)-2,3-butanediol, p-TsOH, toluene, overnight, 93%; (c) nBuLi, THF, −78 °C, 14 h, 47% 15 and 12% 15a; (d) 2 M HCl, THF, 18 h, 95%; (e) TBAF, THF, 0 °C, 15 h, 96%; (f) Pd/BaSO4, H2, MeOH, 19 h, 57%.

Scheme 4.
Synthesis of d9-(R,R)-blumenol B 1. Reagents and conditions; (a) NaH, CD3I, THF, 0 °C—reflux, overnight, 77%; (b) LDA, Et3N, TMS-Cl, THF, −78 °C—r.t., overnight, 82%; (c) 10% Br2 in CH2Cl2, DBU, CH2Cl2. 0 °C, overnight, 85%; (d) 2M HCl, THF, r.t., overnight, quant.; (e) 2R,3R-(-)-2,3-butanediol, p-TsOH, toluene, overnight, 89%; (f) nBuLi, THF, −78 °C, overnight, 51%; (g) 2 M HCl, THF, r.t., 18 h, 94%; (h) TBAF, THF, 0 °C, 15 h, quant.; (i) Pd/BaSO4, H2, MeOH, 8 h, 44%.
Scheme 4.
Synthesis of d9-(R,R)-blumenol B 1. Reagents and conditions; (a) NaH, CD3I, THF, 0 °C—reflux, overnight, 77%; (b) LDA, Et3N, TMS-Cl, THF, −78 °C—r.t., overnight, 82%; (c) 10% Br2 in CH2Cl2, DBU, CH2Cl2. 0 °C, overnight, 85%; (d) 2M HCl, THF, r.t., overnight, quant.; (e) 2R,3R-(-)-2,3-butanediol, p-TsOH, toluene, overnight, 89%; (f) nBuLi, THF, −78 °C, overnight, 51%; (g) 2 M HCl, THF, r.t., 18 h, 94%; (h) TBAF, THF, 0 °C, 15 h, quant.; (i) Pd/BaSO4, H2, MeOH, 8 h, 44%.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, S.M.; Rees, S.W.P.; Jelley, R.E.; Wang, J.; Fedrizzi, B.; Barker, D. Enantioselective Total Synthesis of (R,R)-Blumenol B and d9-(R,R)-Blumenol B. Molecules 2022, 27, 7294. https://doi.org/10.3390/molecules27217294
AMA Style
Tan SM, Rees SWP, Jelley RE, Wang J, Fedrizzi B, Barker D. Enantioselective Total Synthesis of (R,R)-Blumenol B and d9-(R,R)-Blumenol B. Molecules. 2022; 27(21):7294. https://doi.org/10.3390/molecules27217294
Chicago/Turabian StyleTan, Shi Min, Shaun W. P. Rees, Rebecca E. Jelley, Jin Wang, Bruno Fedrizzi, and David Barker. 2022. "Enantioselective Total Synthesis of (R,R)-Blumenol B and d9-(R,R)-Blumenol B" Molecules 27, no. 21: 7294. https://doi.org/10.3390/molecules27217294