
Trifluoromethoxypyrazines: Preparation and Properties


Institute of Organic Chemistry, NAS of Ukraine, Murmans’ka St. 5, Kyiv 02660, Ukraine
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(9), 2226; https://doi.org/10.3390/molecules25092226
Submission received: 30 March 2020
/
Revised: 23 April 2020
/
Accepted: 7 May 2020
/
Published: 9 May 2020
(This article belongs to the Special Issue Organofluorine Chemistry)
Abstract
:The incorporation of the trifluoromethoxy group into organic molecules has become very popular due to the unique properties of the named substituent that has a “pseudohalogen” character, while the chemical properties of the synthesized compound, especially heterocycles with such a group, are less studied. As trifluoromethoxy-substituted pyrazines are still unknown, we have developed efficient and scalable methods for 2-chloro-5-trifluoromethoxypyrazine synthesis, showing the synthetic utility of this molecule for Buchwald-Hartwig amination and the Kumada-Corriu and Suzuki and Sonogashira coupling reactions. Some comparisons of chlorine atom and trifluoromethoxy group stability in these transformations have been carried out.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
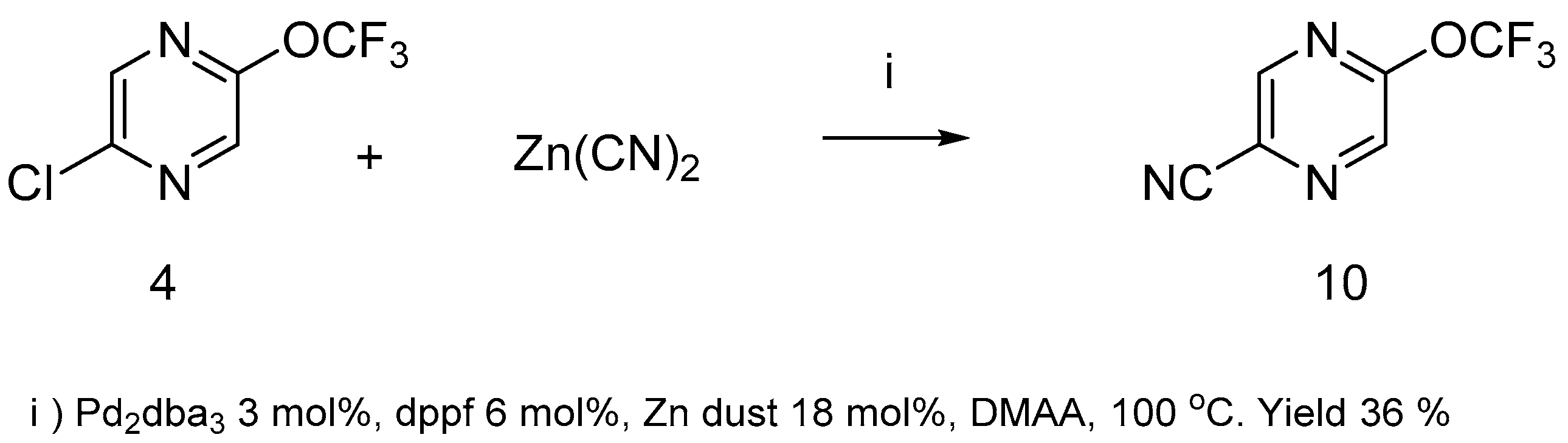{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Among fluorine-containing substituents, the trifluoromethoxy group have gained considerable interest from the modern synthetic audience due to its high influence on molecules for biological and material sciences [1,2]. This moiety is often viewed as a privileged substituent in the field of medicinal chemistry and is routinely considered during drug design [3]. This growing interest in trifluoromethyl ethers is caused by the very peculiar characteristics of the CF3O group. This substituent possesses electron-withdrawing properties by the induction effect and an electron-donating one by the mesomeric for chlorine atoms. For this reason, it has been named “super-halogen” or “pseudo-halogen” [2,4,5]. Moreover, it is one of the most hydrophobic substituents (π(OCF3) = +1.04 compared to π(OCH3) = −0.02) [6]. Additionally, the unique conformation of trifluoromethoxyarenes, in which the OCF3 moiety is orthogonal to the ring plane, must be noted [7,8].
The trifluoromethoxy group attached to aromatics and heteroaromatics is relatively inert and demonstrates the highest stability towards heating, acidic or basic conditions among other fluorine-containing substituents, such as SCF3, CF3, CHF2, OCHF2, SHCF2, and OCH2F [9,10,11,12]. The synthetic utility of pyridine with the OCF3 group was well documented by Leroux and coworkers [8]. It was shown that the trifluoromethoxy group in the 3-rd and even 2-nd position of the ring is tolerant to various reaction conditions, including metallation and catalytic reduction. The authors showed that the trifluoromethoxy group in the 4-th position is less stable and undergoes replacement by bromine atoms under the action of TMSBr. The trifluoromethoxy group easily undergoes nucleophilic displacement when attached to a electron-deficient benzene ring, which was demonstrated by Langlois and coworkers for the example of dinitro-trifluoromethoxybenzene [5].
Taking into account that pyrazine derivatives play an important biological role as food components and scent markers [13,14,15], as well as a number of pharmaceuticals (antitubarcular pyrazinamide, anti-HIV oltipraz, proteasome inhibitor bortezomib, narcotic addiction varenceline, fungal antibiotic aspergillic acid, pulmonary heart disease drug ligustrazine, hypnotic eszopiclone, and eye drops for glaucoma or ocular hypertension brimonidine [13,16]), the trifluoromethoxylated pyrazines family provide perspective molecules for investigation.
Since the first publication in 1955 [17], the main classical methods of trifluoromethoxy ether syntheses are based on chlorination/fluorination techniques or the oxidative desulfofluorination of xanthates [8,18,19,20,21,22]. The range of novel approaches, including the electrophilic trifluoromethylation of oxygen atoms [3,23,24,25,26,27,28] or the incorporation of the trifluoromethoxy group [5,29,30,31,32,33,34], were developed during recent years and successfully applied for aliphatic, aromatic, and heteroaromatic substrates. Nevertheless, pyrazines with such a group are still unknown.
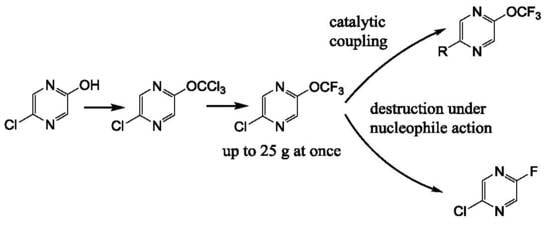
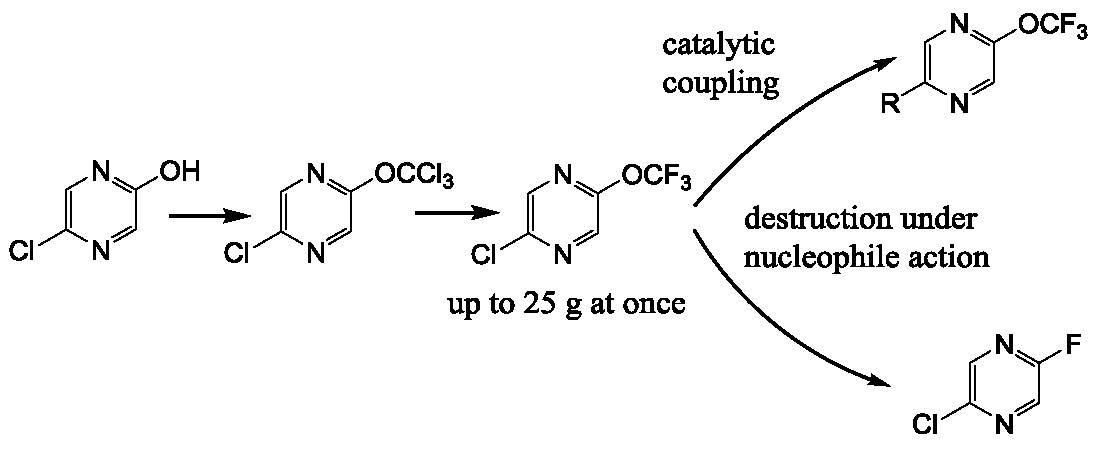
A pyrazine ring is more electron-deficient than a pyridine one due to the influence of the second nitrogen atom [13]. The highly electron-deficient nature of the pyrazine system allows the successful use of the aromatic nucleophilic substitution and cross-coupling reactions for the functionalization of halogenated pyrazines [13,16,35]. 2-Chloro-5-trifluoromethoxypyrazine is a unique compound because both substituents—chlorine atoms and the trifluoromethoxy group—are in equivalent positions. This specific feature opens the possibility to compare the mentioned moieties in various types of reactions and raises the question: are the pyrazines bearing trifluoromethoxy group stable and suitable for synthetic transformations?
2. Results and Discussion
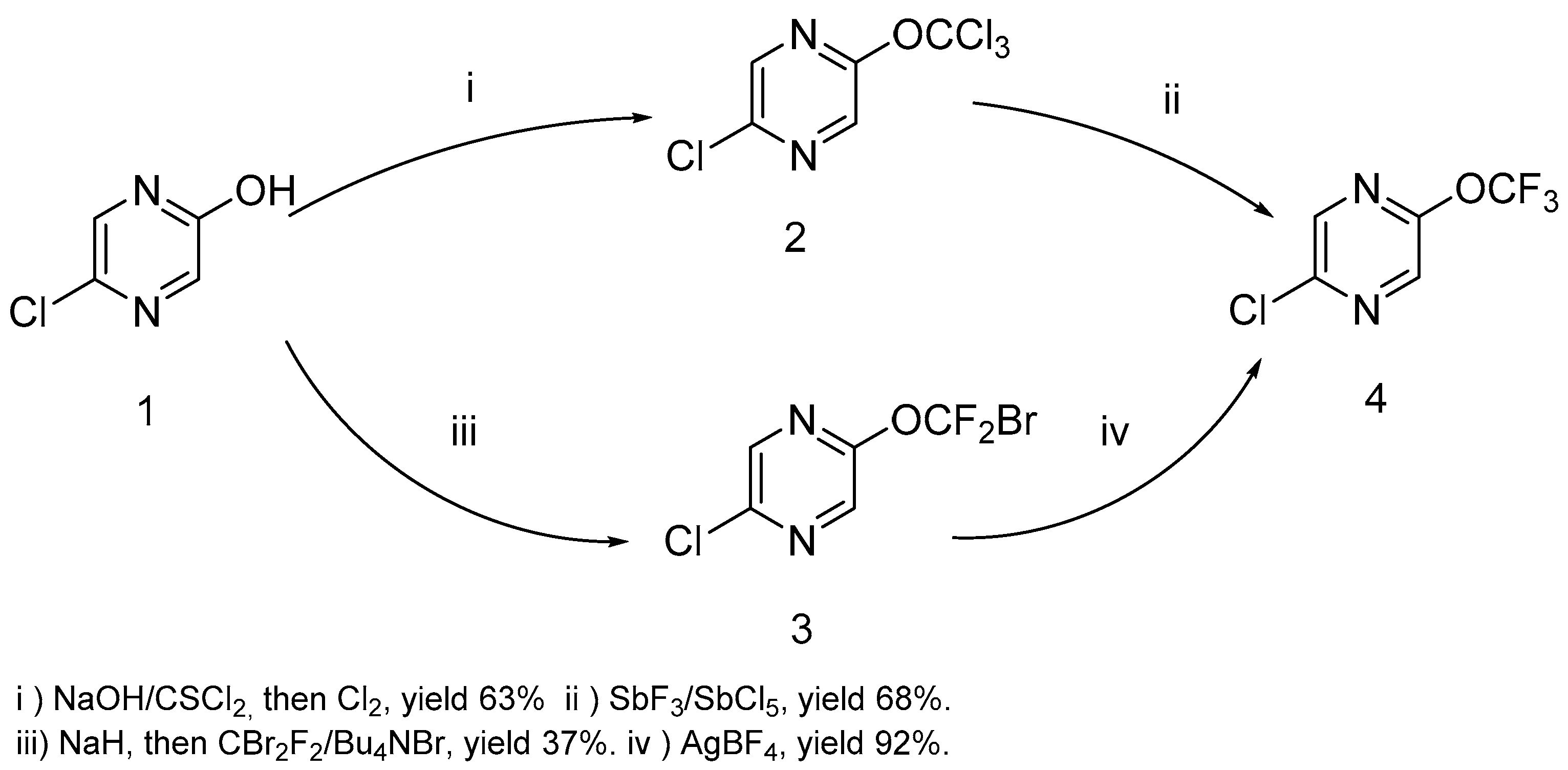
We have prepared 2-chloro-5-trifluoromethoxypyrazine 4 by the action of thiophosgene on hydroxypyrazine, with further chlorination and a chlorine/fluorine exchange using antimony trifluoride (Scheme 1). We used an alternative route based on the halophilic alkylation of pyrazine by dibromodifluoromethane, followed by a bromine/fluorine exchange with silver tetrafluoroborate (Scheme 1). The last method is less favorable due to the low yield in the first stage and the expensive reagents used. Nonetheless, both methods are suitable for a large scale experiment (pyrazine 4 was prepared in 20–25 g scale) in common laboratory glass equipment.
It must be noted that the presence of chlorine atoms in the pyrazine ring is critical for the success of both methods. We have found that trichloromethoxy and bromodifluoromethoxypyrazine cannot be prepared by these methods starting from 2-hydroxypyrazine. The same peculiarity was shown by Leroux and coworkers for a trifluoromethoxypyridine preparation [8].
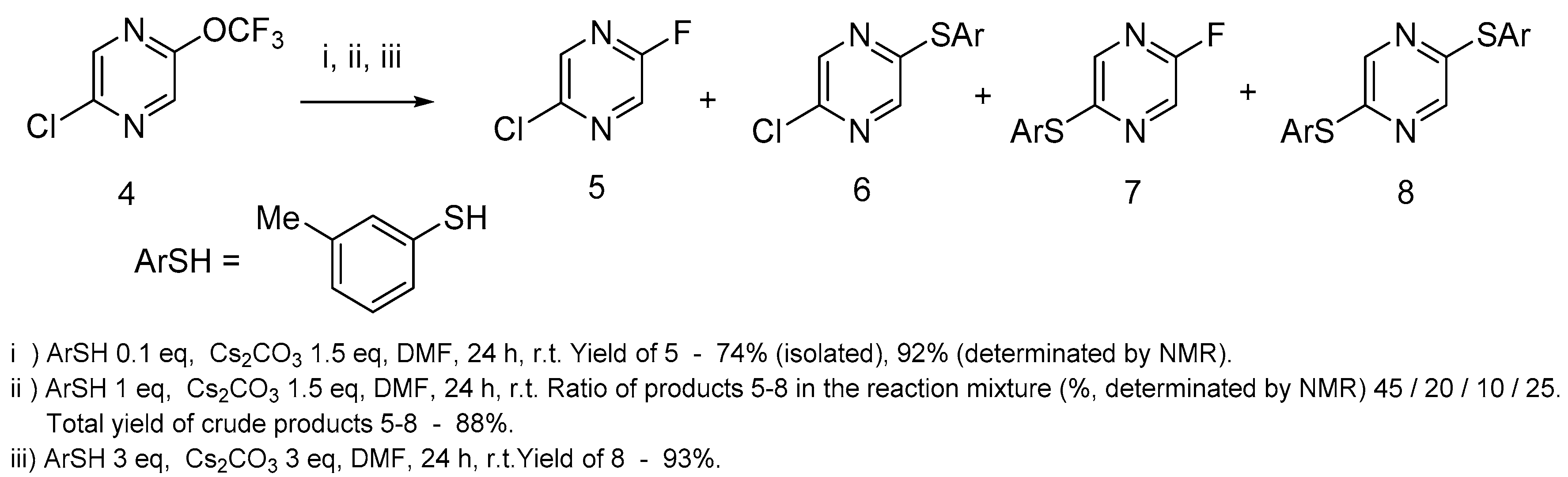
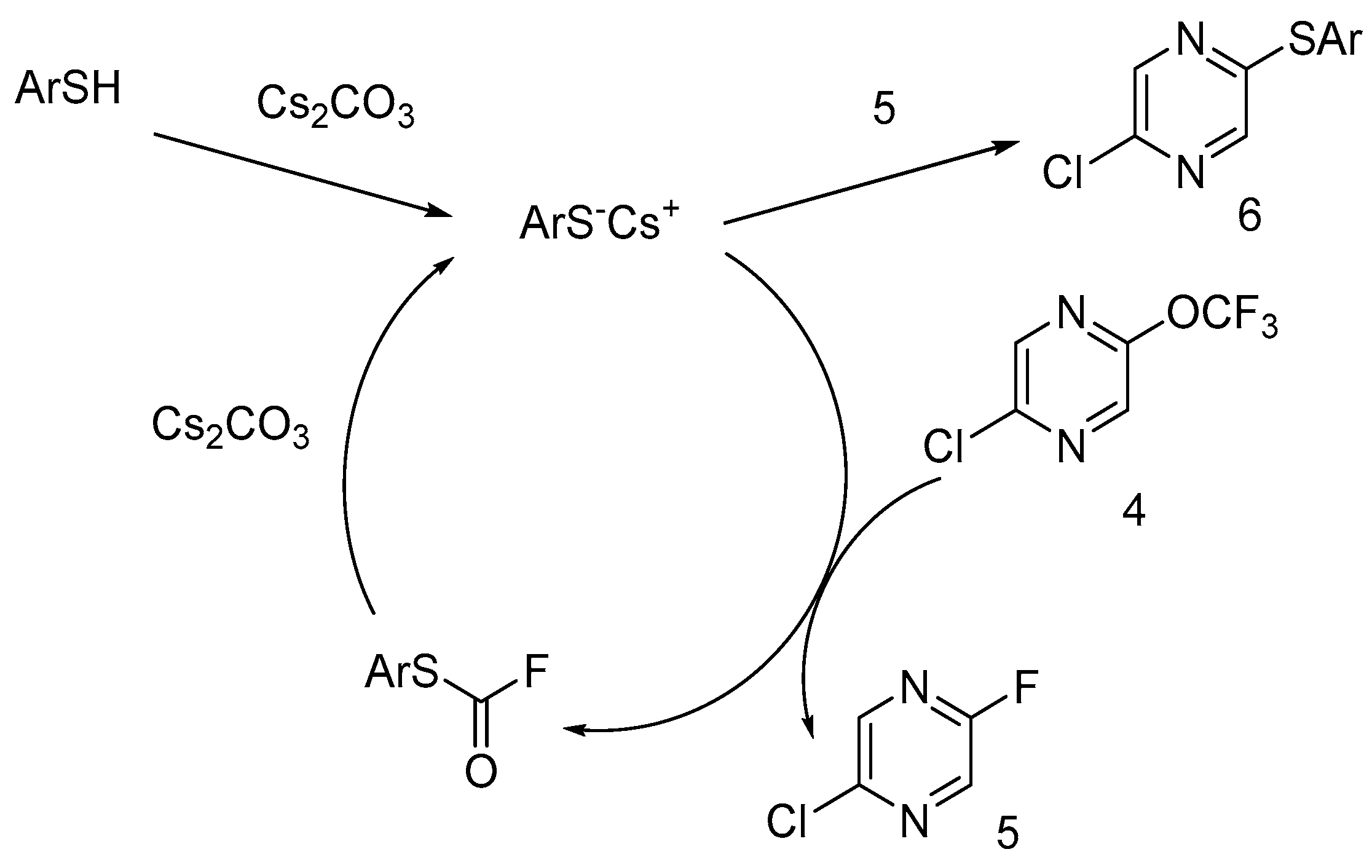
We have studied the 2-chloro-5-trifluoromethoxypyrazine behavior with nucleophiles using 3-methylthiophenol as an example of S-nucleophile. If one equivalent of 3-methylthiophenol was used in this reaction, 2-chloro-5-fluoropyrazine 5 was the main fluorinated product (Scheme 2), but a range of products with thiophenol moiety were also formed in these conditions. Surprisingly, 2-chloro-5-fluoropyrazine 5 was the main product when pyrazine 4 reacted with 10 mol% 3-methylthiophenol and an excess of cesium carbonate at room temperature. In the absence of thiophenol, this reaction did not occur even after 1 week of stirring at r.t. Pyrazine 8 was the single product when this reaction was performed with an excess of 3-methylthiophenol.
Taking into account the destruction of OCF3 moiety in the presence of catalytic amounts of thiophenolate anions, we proposed the following scheme for trifluorometoxy group degradation (Scheme 3): the thiophenolate anion reacts with trifluorometoxy pyrazine 4, with an S-aryl fluorocarbamate formation that can be recovered to thiophenolate by the action of cesium carbonate. The nucleophilic substitution of the fluorine atom of pyrazins 5 and 7 or the chlorine atom of pyrazines 5 and 6 is the route of removing thiophenol from the catalytic cycle (only pyrazine 5 was shown in Scheme 3 as an example). A high yield of fluoropyrazine 5 is evidence of the high catalytic activity of thiophenol for this reaction.
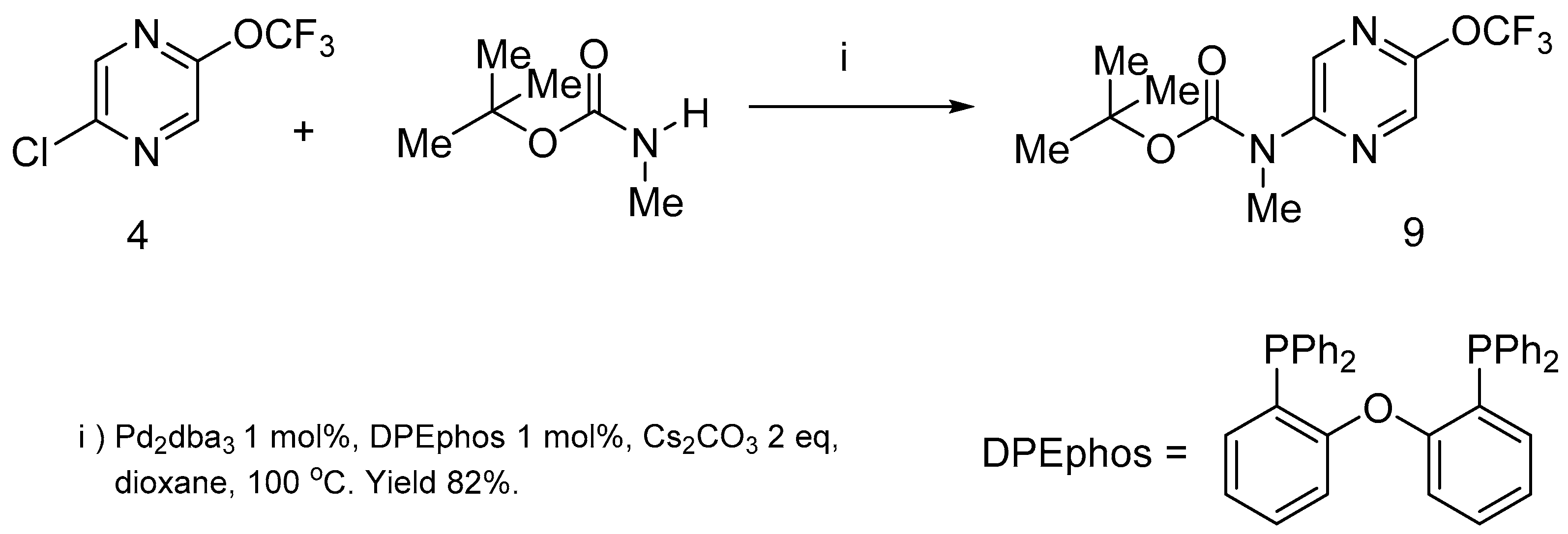
The nucleophilic substitution of chlorine atoms in pyrazine 4 by diethylamine as an example of N- nucleophiles did not occur at room temperature. The prolonged heating (24 h) of the reaction mixture in various solvents at 100 °C led to the complete destruction of the starting material. Nevertheless, the chlorine atom of this molecule was successfully replaced by nitrogen nucleophile under Buchwald-Hartwig amination reaction conditions [35,36,37]. Palladium dibenzoylacetonate (Pd2dba3)/DPEphos catalysis took place and the Boc-protected amine 9 was prepared in high yield (Scheme 4).
The replacement of chlorine atom in compound 4 with the nitrile group using potassium cyanide failed, however pirazinenitrile 10 was prepared with zinc cyanide under Pd2dba3/diphenylphosphinoferrocene (dppf) catalysis (Scheme 5).
Alkylpyrazines, especially bearing methoxy group ones, are compounds of great interest due to their odor properties [38,39,40]. For instance, the odor threshold of 2-methoxy-3-sec-buthyl-pyrazine in water solution is 1 part per 1012 parts of water [39].
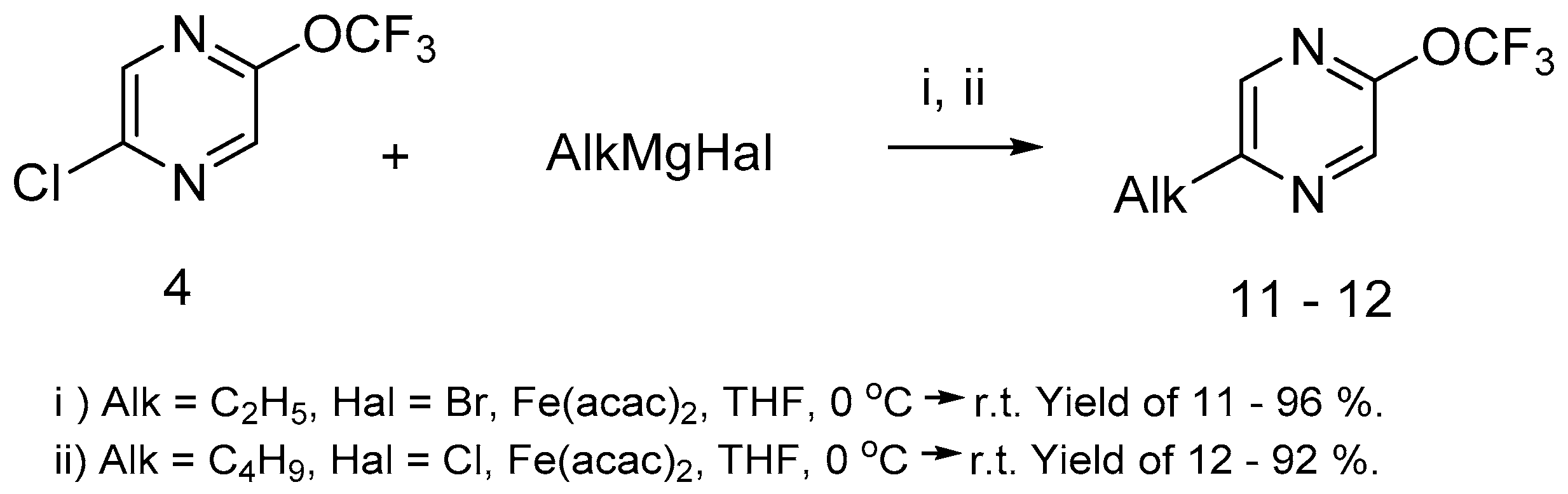
We have shown that the alkyl pirazines-bearing trifluoromethoxy group can be prepared in high yield by Kumada–Corriu coupling [41,42,43] with iron acetylacetonate (Fe(acac)2) as a catalyst (Scheme 6). It must be noted that both alkylmagnesium halides (bromide or chloride) are suitable for this reaction.
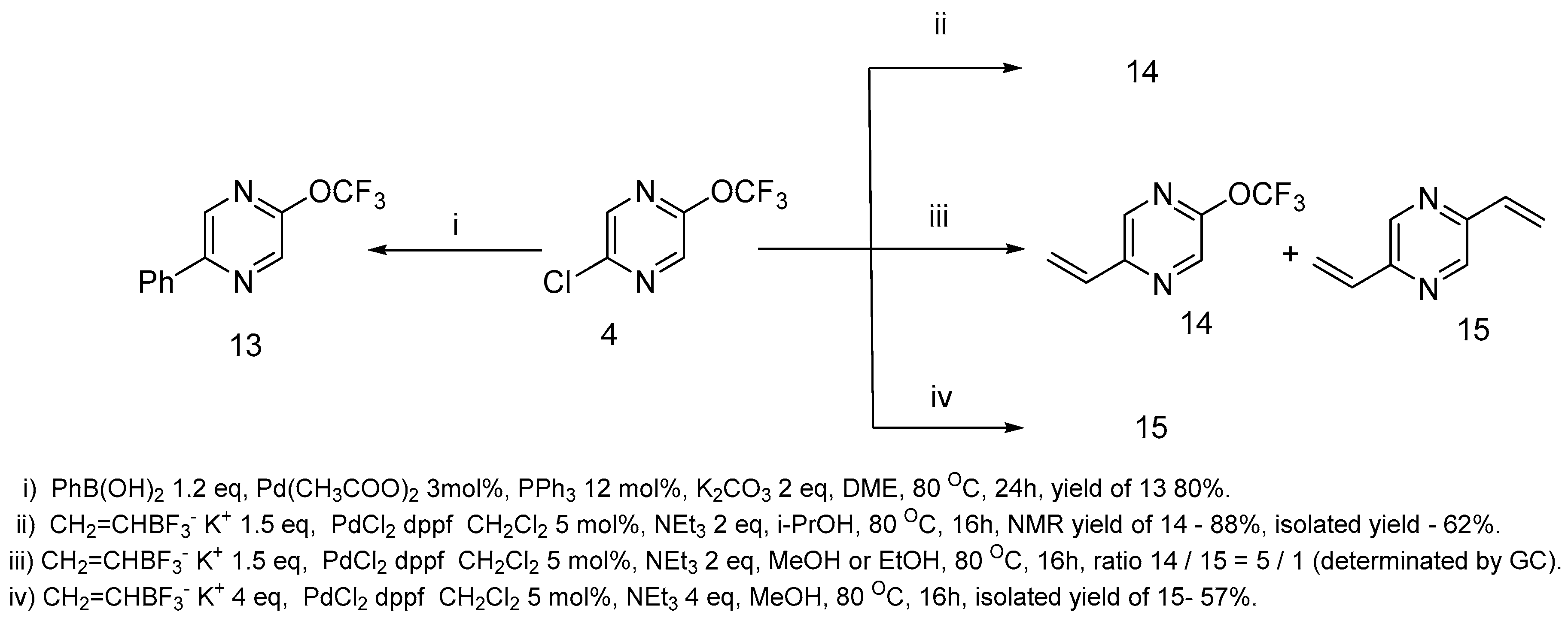
Suzuki coupling [44] is a powerful method for pyrazine derivatization [35]. We have found that 2-chloro-5-trifluoromethoxypyrazine 4 is a suitable substrate for coupling with boronic acids as well as with trifluoroborates. The reaction with phenylboronic acid resulted in phenylpyrazine 13 formation in a high yield (Scheme 7). In the case of potassium ethenyl trifluoroborate, the result of the reaction is dependent on the solvents used (Scheme 7). We tested various solvents for this reaction (DMF, MeOH, EtOH, i-PrOH). In DMF, unidentified products were formed. Meanwhile, in the cases of MeOH and EtOH, the reaction resulted in the substitution of the chlorine atom to give product 14, as well as both the chlorine atom and trifluoromethoxy group removal with the pyrazine 15 formation. Bis-ethenyl pyrazine 15 was formed selectively if a large excess of potassium ethenyl trifluoroborate was used. Monoethenyl pyrazine 14 was produced selectively when i-PrOH was used as the solvent. Unfortunately, it is difficult to isolate the product in a pure state in this case, and pyrazine 14 was obtained only with a 62% yield.
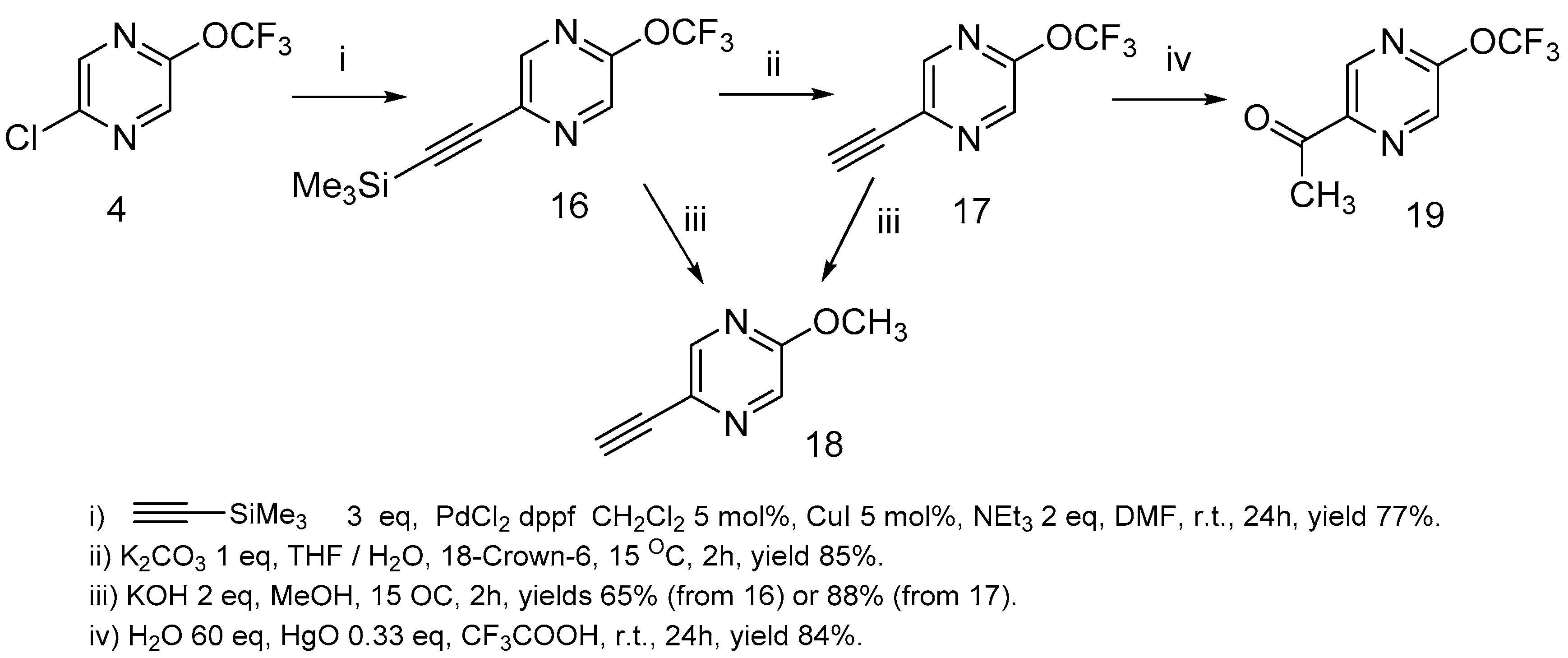
The reaction of 2-chloro-5-trifluoromethoxypyrazine 4 with ethynyltrimethylsilane under Sonogashira [45] reaction conditions led to 2-ethynyl-5-trifluoromethoxypyrazine 16 formation in a high yield (Scheme 8).
Ethynylpyrazine 16 can be successfully desililated by the action of potassium carbonate under heterogeneous reaction conditions in the THF/water mixture. When the desililation of pyrazine 16 was performed with KOH in a methanol/water solution, as was described in the literature [46], a mixture of trifluoromethoxypirazine 17 and methoxypyrazine 18 was obtained. Methoxypyrazine 18 was prepared starting from trifluoromethoxypyrazine 17 or sililated pyrazine 16 by the action of KOH in methanol. The hydration of acetylene 17 via the Kucherov reaction [47,48] resulted in 2-acethyl-5-trifluoromethoxy-pyrazine 19 formation in a high yield (Scheme 8).
3. Materials and Methods
1H-NMR spectra were recorded at 300 MHz with a Varian VXR-300 spectrometer (Varian Inc., Palo Alto, CA, USA), at 500 MHz with a Bruker AVANCE DRX 500 instrument (Bruker, Billerica, MA, USA), or at 400 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA).13C-NMR-spectra (proton decoupled) were recorded on a Bruker AVANCE DRX 500 instrument at 125 MHz, or at 100 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA). 19F-NMR spectra were recorded at 376 MHz with a Varian UNITY-Plus 400 spectrometer (Varian Inc., Palo Alto, CA, USA). The chemical shifts are given in ppm relative to Me4Si and CCl3F, respectively, as internal or external standards. The 1H-, 13C-, and 19F-NMR spectra of the compounds can be found in the Supplementary Materials. The LC-MS spectra were registered on an “Agilent 1100 Series” instrument with a diode-matrix and mass-selective detector “Agilent 1100 LS/MSD SL” (ionization method: chemical ionization at atmospheric pressure; ionization chamber operation conditions: simultaneous scanning of positive and negative ions in the range 80–1000 m/z, Agilent Technologies, Santa Clara, CA, USA). The GC-MS spectra were registered on a Hewlett-Packard HP GC/MS 5890/5972 instrument (EI 70 eV) (Philips, Bothell, WA, USA). The melting points were determined in open capillaries using an SMP3 instrument (Stuart Scientific Bibby Sterlin Ltd., Stone, Staffordshire, UK). An elemental analysis was performed in the Analytical Laboratory of the Institute of Organic Chemistry, NAS, Ukraine, Kyiv.
Unless otherwise stated, commercially-available reagents were purchased from Enamine Ltd. (Kyiv, Ukraine) and were used without purification. The solvents were purified according to the standard procedures [49]. Antimony trifluoride was sublimated immediately prior to use. The 2-Hydroxy-5-chloropyrazine 1 was prepared starting from 2-amino-5-chloropyrazine according to method [50] in a 37 g scale.
All the reactions were performed in an argon atmosphere. For the column chromatography, Merck Kieselgel 60 silica gel (Merck, Darmstadt, Germany) was used. Thin-layer chromatography (TLC) was carried out on aluminum-backed plates coated with silica gel (Merck Kieselgel 60 F254, Merck, Darmstadt, Germany).
3.1. 2-Chloro-5-(trichloromethoxy)pyrazine 2
5-Chloropyrazin-2-ol 1 (40 g, 0.31 mol) was added in portions to the solution of NaOH (14.1 g, 0.35 mol) in water (250 mL) at 5–10 °C. The mixture was cooled to 0 °C and the solution of thiophosgene (35.3 g, 0.31 mol) in chloroform (250 mL) was added dropwise over 1 h under vigorous stirring. After the addition was complete, the mixture was vigorously stirred for a further 3 h at 0 °C before being extracted with chloroform (5 × 100 mL). The combined organic layers were washed with water and dried with MgSO4. After the dehydration agent had been filtered off, the mixture was saturated with chlorine and stirred for 3 days at 25 °C. The excess of chlorine was removed with nitrogen gas stream, the solvent was distilled off in a vacuum (300 mbar), and the residue was distilled in a vacuum, yielding trichloromethoxypyrazine 2 as a colorless oil or solid. The yield was 47.9 g (63%); b.p. 100–101 °C (1 mbar); m.p. 26–28 °C. 1H-NMR (400 MHz, CDCl3) δ 8.32 (s, 1H, HPy), 8.37 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 108.9 (s, OCCl3), 136.0, 140.8, 145.6, 154.0. GC-MS, 70 eV, m/z (rel. int.): 248 (100) [M(335Cl + 37Cl)]+, 246 (80) [M(435Cl)]+, 250 (50) [M(235Cl + 237Cl)]+. Anal. Calcd. for C4H2ClFN2: Cl, 57.20; Found: Cl, 57.26.
3.2. 2-(Bromodifluoromethoxy)-5-chloropyrazine 3
To the solution of 5-chloropyrazin-2-ol (40 g, 0.31 mol) and tetrabutylammonium bromide (4.9 g, 15 mmol) in anhydrous DMF (300 mL), NaH (60% in mineral oil) (14.7 g, 0.37 mol) was added in portions at 0 °C and the mixture was stirred for 2 h at r.t. The mixture was cooled to −30 °C and dibromodifluoromethane (257 g, 1.23 mol) was added dropwise. The mixture was carefully warmed and stirred at 30–35 °C for 6 h. Water (900 mL) was added dropwise to the mixture and the excess of CBr2F2 was distilled in a dry ice trap. The residue was extracted with MTBE (tert-butyl-methyl ether) (7 × 100 mL), extract was washed with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum. The yield was 29.4 g (37%); colorless liquid or solid; b.p. 80–81 °C (10 mbar); m.p. 14–17 °C. 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, HPy), 8.35 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 111.5 (t, 1JCF = 315.5 Hz, CBrF2), 135.2, 141.0, 145.9, 152.6. 19F-NMR (376.5 MHz, CDCl3) δ −18.4 (s, CBrF2). GC-MS, 70 eV, m/z (rel. int.): 258 (75) [M(79Br + 35Cl)]+, 260 (100) [M(79Br + 37Cl) or (81Br + 35Cl)]+, 262 (25) [M(81Br + 37Cl)]+. Anal. Calcd. for C5H2BrClF2N2O: C, 23.15; H, 0.78; N, 10.80; Found: C, 23.12; H, 0.68; N, 10.72.
3.3. 2-Chloro-5-trifluoromethoxypyrazine 4
Method A. The mixture of freshly sublimated SbF3 (103.7 g, 0.58 mol) and SbCl5 (17.4 g, 0.06 mol) was heated at 125–130 °C for 15 min and 2-chloro-5-(trichloromethoxy)pyrazine 2 (47.9 g, 0.19 mol) was added in portions at 100 °C to this mixture. The reaction mixture was stirred at 145–150 °C for 5 h and then 1 h at 155–160 °C. After cooling to r.t., the mixture was suspended in CH2Cl2 (700 mL), and solutions of K2CO3 (138 g, 1 mol) in 700 mL of water and KF (174 g, 3 mol) in 500 mL of water were carefully added to the mixture. The mixture was filtered through celite, the organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
Method B. Silver tetrafluoroborate (24.3 g, 0.125 mol) was added in portions to the solution of 2-(bromodifluoromethoxy)-5-chloropyrazine 3 (29.4 g, 0.11 mol) in CH2Cl2 (250 mL) at −78 °C. The mixture was warmed to r.t. and stirred for 24 h in darkness. The solution of sodium bicarbonate (11.4 g, 0.136 mol) in 150 mL of water was added to the reaction mixture and the mixture was filtered through celite. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
The yield (Method A) was 26.0 g (68%); (Method B) 20.7 g (92%); colorless liquid; b.p. 80–81 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 8.23 (s, 1H, HPy), 8.29 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 119.5 (q, 1JCF = 262.5 Hz, CF3), 134.6, 140.9, 145.8, 151.8. 19F-NMR (376.5 MHz, CDCl3) δ −57.6 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 198 (100) [M(35Cl)]+, 20 (30) [37Cl)]+. Anal. Calcd. for C5H2ClF3N2O: C, 30.25; H, 1.02; Cl, 17.86; N, 14.11; Found: C, 30.22; H, 0.98; Cl, 17.93; N, 14.09.
3.4. Reaction of 2-Chloro-5-trifluoromethoxypyrazine 4 with 3-Methylthiophenol and Alternative Route to 2-Chloro-5-fluoropyrazine 5
Method A. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.0 g 5 mmol), 3-methylthiophenol (0.06 g, 0.5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
Method B. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.0 g 5 mmol), 3-methylthiophenol (0.62 g, 5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the volatile products were distilled into a liquid nitrogen trap in a high vacuum (0.2 mbar) at 35–40 °C (bath temperature). After redistillation at 100 mbar, 2-chloro-5-fluoropyrazine was obtained. The residue after the high vacuum distillation was separated by TLS chromatography using pentane/ethylacetate (5:1 by vol.) as an eluent, yielding a mixture of pirazines 6 and 7 (with ratio 1:2 by weight) and disubstituted pyrazine 8.
Method C. The mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (0.5 g 2.5 mmol), 3-methylthiophenol (0.93 g, 7.5 mmol), and cesium carbonate (2.46 g, 7.5 mmol) in anhydrous DMF (5 mL) was stirred at r.t. under Argon for 24 h. The mixture was poured into water (20 mL) and extracted with ether (5 × 10 mL). The etheral solution was washed with 5% sodium bicarbonate solution and then with brine and dried with MgSO4. The solvent was distilled off in a vacuum and the residue was purified by column chromatography using a mixture of hexane/ethylacetate (5:1 by vol.) as an eluent, yielding disubstituted pyrazine 8.
An alternative route to 2-chloro-5-fluoropyrazine5. To the solution of 2-amino-5-chloropyrazine (5 g, 38.6 mmol) in 50% aqueous HBF4 (20 mL), sodium nitrite (5.3 g, 77.2 mmol) was added in portions at 0–5 °C. The reaction mixture was gently heated to 18–20 °C and stirred for 4 h. The mixture was extracted with ether (5 × 20 mL), and the etheral solution was washed with brine and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
2-Chloro-5-fluoropyrazine5. The yield was (Method A) 0.49 (74%), (method B) 0.12 g (18%), (alternative method) 4.77 g (93%); colorless liquid, b.p. 80–81 °C (100 mbar). 1H-NMR (500 MHz, CDCl3) δ 8.22 (s, 1H, H3′), 8.24 (d, 1H, 3JHF = 8.5 Hz, H6′). 13C-NMR (126 MHz, CDCl3) δ 132.4 (d, 2JCF = 40.2 Hz, C6′), 141.2 (d, 3JCF = 10.0 Hz, C3′), 145.3 (CCl), 159.3 (d, 1JCF = 252.7 Hz, CF). 19F-NMR (376.5 MHz, CDCl3) δ −85 (s, CF). GC-MS, 70 eV, m/z (rel. int.): 132 (100) [M(35Cl)]+, 134 (32) [M(37Cl)]+. Anal. Calcd. for C4H2ClFN2: C, 36.25; H, 1.52; Cl, 26.75; N, 21.14; Found: C, 36.22; H, 1.53; Cl, 26.73; N, 21.09.
The mixture of 2-chloro-5-(m-tolylthio)pyrazine6and 2-fluoro-5(m-tolylthio)pyrazine7. The yield of the mixture was 0.33 g: chloropyrazine 6–11%, fluoropyrazine 7–22% (the yield was determined by NMR and LC analyses of the mixture of pyrazines 6 and 7). Rf = 0.8 pentane/ethylacetate (5:1). Chloropyrazine 6: 1H-NMR (500 MHz, CDCl3) δ 2.36 (s, 3H, CH3), 7.25–7.27 (m, 1H, CHAr), 7.34–7.37 (m, 1H, CHAr), 7.40–7.42 (m, 2H, 2CHAr), 7.96 (s, 1H, H6′Py), 8.37 (s, 1H, H6′Py); LC/MS (CI): m/z = 237 [M(35Cl) + H]+, 239 [M(37Cl) + H]+. Fluoropyrazine 7 1H-NMR (500 MHz, CDCl3) δ 2.36 (s, 3H, CH3), 7.25–7.27 (m, 1H, CHAr), 7.34–7.37 (m, 1H, CHAr), 7.40–7.42 (m, 2H, 2CHAr), 7.84 (s, 1H, H6′Py), 7.26 (d, 1H, 3JHF = 8.5 Hz, H3′Py). 19F-NMR (376.5 MHz, CDCl3) δ −87.9 (s, CF). LC/MS (CI): m/z = 221 [M + H]+.
2,5-Bis(m-tolylthio)pyrazine8. Yield (method B) 0.4 g (25%), (method C) 0.75 g (93%); yellowish powder, m.p. 102–103 °C. Rf = 0.6 pentane/ethylacetate (5:1). 1H-NMR (400 MHz, CDCl3) δ 2.34 (s, 6H, 2CH3), 7.18–7.21 (m, 2H, 2CHAr), 7.27 (t, 2H, 3JHH = 7.6 Hz, 2m-CHAr), 7.33–7.37 (m, 4H, 4CHAr), 8.03 (s, 2H, CHPy). 13C-NMR (126 MHz, CDCl3) δ 21.3, 129.4, 129.6, 130.3, 131.7, 135.2, 139.7, 142.5, 153.6. LC/MS (CI): m/z = 325 [M + H]+. Calcd. for C18H16N2S2: C, 66.63; H, 4.97; N, 8.63; S, 19.76; Found: C, 66.68; H, 5.02; N, 8.59; S, 19.70.
3.5. tert-Butyl Methyl(5-(trifluoromethoxy)pyrazin-2-yl)carbamate 9
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2 g, 10 mmol), N-Boc-methylamine (2 g, 15 mmol) and Cs2CO3 (6.5 g, 20 mmol) in anhydrous dioxane (30 mL) Pd2dba3 (0.09 g, 0.1 mmol) and bis[(2-diphenylphosphino)phenyl] ether (DPEphos) (0.05 g, 0.1 mmol) were added and the mixture was stirred at 100 °C for 12 h. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with ethylacetate (5 × 10 mL). The organic extract was washed with brine, dried with MgSO4, and concentrated in a vacuum. After column chromatography with the mixture of hexane/ethylacetate (5:1 by vol.) as the eluent, pyrazine 9 was obtained. The yield was 2.4 g (82%); colorless powder, m.p. 78–79 °C. 1H-NMR (400 MHz, CDCl3) δ 1.54 (s, 9H, C(CH3)3), 3.39 (s, 3H, CH3), 8.18 (s, 1H, HPy), 8.82 (s, 1H, HPy). 13C-NMR (100 MHz, CDCl3) δ 28.2 (C(CH3)3), 33.8 (NCH3), 82.5 (C(CH3)3), 120.2 (q, 1JCF = 262.5 Hz, CF3), 132.1, 137.6, 148.5, 150.1, 153.6. 19F-NMR (376.5 MHz, CDCl3) δ −57.5 (s, CF3). LC/MS (CI): m/z = 294 [M + H]+. Calcd. for C11H14F3N3O3: C, 45.05; H, 4.81; N, 14.33; Found: C, 45.08; H, 4.88; N, 14.29.
3.6. 5-(Trifluoromethoxy)pyrazine-2-carbonitrile 10
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (1.00 g, 5 mmol), Zn(CN)2 (0.39 g, 3.3 mmol) and Zn dust (0.6 g, 0.9 mmol) in anhydrous dimethylacetamide (10 mL) Pd2dba3 (0.14 g, 0.15 mmol) and dppf (0.17 g, 0.3 mmol) were added, and the mixture was stirred at 100 °C for 8 h. After cooling to r.t., the mixture was diluted with water (30 mL), filtered through celite, and extracted with ether (7 × 10 mL). The etheral solution was washed with brine (3 × 10 mL), dried with MgSO4, and the solvent was distilled off under atmospheric pressure. The residue was purified by TLC chromatography, yielding 0.34 g (36%) cyanopyrazine 10 as a colorless liquid with an intense almond scent, b.p. 95–96 °C (100 mbar). Rf = 0.8 pentane/ethylacetate (10:1). 1H-NMR (400 MHz, CDCl3) δ 8.51 (s, 1H, HPy), 8.62 (s, 1H, HPy). 13C-NMR (125.6 MHz, CDCl3) δ 114.5 (CN), 119.8 (q, 1JCF = 266.5 Hz, CF3), 127.5, 136.9, 145.7, 154.3. 19F-NMR (376.5 MHz, CDCl3) δ −57.5 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 189 (100) [M]+. Calcd. for C6H2F3N3O: C, 38.11; H, 1.07; N, 22.22; Found: C, 38.18; H, 1.16; N, 22.19.
3.7. Synthesis of Alkylpyrazines-Bearing Trifluoromethoxy Group by Kumada-Corriu Coupling
The solution of ethylmagnesium bromide (18 mL, 18 mmol, 1 mol/L solution in THF) or butylmagnesium chloride (9 mL, 18 mmol, 2 mol/L solution in THF) was added dropwise at 0 °C in an Argon atmosphere to the mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (3.0 g, 15 mmol) and Fe(acac)2 (0.19 g, 0.75 mmol) in 30 mL of anhydrous THF. The mixture was warmed to r.t. and stirred for 3 h (in the case of ethylmagnesium bromide) or 4 h (in the case of butylmagnesium chloride). The reaction mixture was cooled to 0 °C and neutralized with 3% aqueous HCl. The product was extracted with ether (5 × 10 mL) and the etheral solution was washed with brine (5 × 10 mL) and dried with MgSO4. The solvent was distilled off at atmospheric pressure and the product was distilled in a vacuum.
2-Ethyl-5-(trifluoromethoxy)pyrazine11. Yield 2.77 g (96%); colorless liquid with intense fruit scent, b.p. 83–85 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 1.31 (t, 3H, 3JHH = 7.6 Hz, CH3), 2.84 (q, 2H, 3JHH = 7.6 Hz, CH2) 8.13 (s, 1H, HPy), 8.35 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 13.3 (s, CH3), 22.8 (s, CH2), 120.1 (q, 1JCF = 262.7 Hz, CF3), 135.0, 140.1, 151.8, 156.7. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 192 (100) [M]+. Anal. Calcd. for C7H7F3N2O: C, 43.76; H, 3.67; N, 14.58; Found: C, 43.62; H, 3.53; N, 14.49.
2-Butyl-5-(trifluoromethoxy)pyrazine12. 3.03 g (92%); colorless liquid with intense fruit scent, b.p. 83–85 °C (20 mbar). 1H-NMR (400 MHz, CDCl3) δ 0.92 (t, 3H, 3JHH = 7.6 Hz, CH3), 1.37 (sext, 2H, 3JHH = 7.6 Hz, CH2CH3), 1.67 (quint, 2H, 3JHH = 7.6 Hz, CH2CH2CH2), 2.82 (t, 2H, 3JHH = 7.6 Hz, CH2) 8.11 (s, 1H, HPy), 8.34 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 13.8 (s, CH3), 22.2 (s, CH2), 31.5 (s, CH2), 34.2 (s, CH2), 120.1 (q, 1JCF = 262.3 Hz, CF3), 135.0, 140.5, 151.8, 155.8. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 220 (100) [M]+. Anal. Calcd. for C9H11F3N2O: C, 49.09; H, 5.04; N, 12.72; Found: C, 49.02; H, 5.03; N, 12.79.
3.8. 2-Phenyl-5-(trifluoromethoxy)pyrazine 13
Palladium acetate (34 mg, 0.15 mmol) and triphenylphosphine (158 mg, 0.6 mmol) were mixed together in carefully degassed dimethoxiethane (25 mL) and stirred for 15 min at r.t. under an Ar atmosphere. Chloropyrazine 4 (1.0 g, 5 mmol), phenylboronic acid (0.7 g, 6 mmol), and K2CO3 (1.4 g, 10 mmol) were added to the reaction mixture and the mixture was stirred at 75 °C for 20 h. After cooling to r.t., the mixture was diluted with water, filtered through celite, and extracted with tret-buthylmethyl ether (3 × 25 mL). The organic solution was washed with brine, dried with MgSO4, and evaporated in a vacuum. The residue was purified by column chromatography with hexane/ethylacetate (10:1 by vol.) as the eluent (Rf = 0.6). The yield was 0.95 g (80%); colorless powder, m.p. 80 °C. 1H-NMR (400 MHz, CDCl3) δ 7.48–7.55 (m, 3H, 3CHPh), 7.92–8.00 (m, 2H, 2CHPh), 8.50 (s, 1H, HPy), 8.70 (s, 1H, HPy). 13C-NMR (126 MHz, CDCl3) δ 120.2 (q, 1JCF = 262.7 Hz, CF3), 126.8, 129.1, 130.0, 135.1, 135.2, 138.4, 150.9, 152.4. 19F-NMR (376.5 MHz, CDCl3) δ −57.2 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 240 (100) [M]+. Anal. Calcd. for C11H7F3N2O: C, 55.01; H, 2.94; N, 11.66; Found: C, 49.9; H, 3.00; N, 11.69.
3.9. Reaction of 2-Chloro-5-trifluoromethoxypyrazine 4 with Potassium Ethenyl Trifluoroborate
3.9.1. 2-(Trifluoromethoxy)-5-vinylpyrazine 14
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2.0 g, 10 mmol), potassium ethenyl trifluoroborate (2.0 g, 15 mmol) and NEt3 (2 g, 20 mmol) in anhydrous iso-propanole (10 mL) PdCl2dppf·CH2Cl2 (0.4 g, 0.5 mmol) were added and the mixture was stirred for 20 h at 75 °C. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with CH2Cl2 (5 × 10 mL). The organic extract was washed with 1% aqueous HCl, then with water, and dried with CaCl2. After the removal of the solvent at atmospheric pressure, the residue was distilled with deflegmator in a vacuum. The yield was 1.17 g (62%); colorless liquid with a pungent odor, b.p. 90–91 °C (100 mbar). 1H-NMR (500 MHz, CDCl3) δ 5.63 (d, 1H, 3JHH = 11.0 Hz, =CH2), 6.33 (d, 1H, 3JHH = 17.5 Hz, =CH2), 6.83 (dd, 1H, 3JHH-trans = 17.5 Hz, 3JHH-cis = 11.0 Hz, =CH2), 8.27 (s, 1H, HPy), 8.41 (s, 1H, HPy). 13C-NMR (100 MHz, CDCl3) δ 119.9 (q, 1JCF = 262.7 Hz, CF3), 120.6, 131.7, 135.1, 139.1, 148.9, 152.1. 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 190 (100) [M]+. Anal. Calcd. for C7H5F3N2O: C, 44.22; H, 2.65; N, 14.73; Found: C, 44.29; H, 2.60; N, 14.69.
3.9.2. 5-Divinylpyrazine 15
The reaction was performed in the glass high pressure tube 50 mL vol. To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (2.0 g, 10 mmol), potassium ethenyl trifluoroborate (5.4 g, 40 mmol) and NEt3 (4.1 g, 40 mmol) in anhydrous methanol (10 mL) PdCl2dppf·CH2Cl2 (0.8 g, 15 mmol) were added and the mixture was stirred for 20 h at 75 °C. After cooling to r.t., the mixture was diluted with water (20 mL), filtered through celite, and extracted with CH2Cl2 (5 × 10 mL). The organic extract was washed with 1% aqueous HCl, then with water, and dried with CaCl2. After the removal of the solvent at atmospheric pressure, the residue was purified by flash column chromatography with pentane/ethylacetate (5:1 by vol.) as the eluent (Rf = 0.8). The yield was 0.75 g (57%); colorless liquid, b.p. 45–47 °C (10 mbar). Compound 15 was partly polymerized under distillation and completely polymerized after 1-month storage in a refrigerator. 1H-NMR (500 MHz, CDCl3) δ 5.60 (d, 2H, 3JHH = 11.0 Hz, 2 =CH2), 6.34 (d, 2H, 3JHH = 17.5 Hz, =CH2), 6.83 (dd, 2H, 3JHH-trans = 17.5 Hz, 3JHH-cis =11.0 Hz, =CH2), 8.54 (s, 2H, HPy). 13C-NMR (100 MHz, CDCl3) δ 120.3, 133.3, 142.4, 149.5. GC-MS, 70 eV, m/z (rel. int.): 132 (100) [M]+. Anal. Calcd. for C8H8N2: C, 72.70; H, 6.10; N, 21.21; Found: C, 72.72; H, 6.10; N, 21.19.
3.10. 2-(Trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16
To the carefully degassed mixture of 2-chloro-5-trifluoromethoxypyrazine 4 (3.0 g, 15 mmol), ethynyltrimethylsilane (4.4 g, 45 mmol) and NEt3 (9.2 g, 90 mmol) in anhydrous DMF (25 mL) PdCl2dppf·CH2Cl2 (0.62 g, 0.75 mmol) and CuI (0.14 g, 0.75 mmol) were added and the mixture was stirred for 20 h at r.t. The mixture was diluted with water (75 mL), filtered through celite, and extracted with MTBE (5 × 50 mL). The organic extract was washed with 1% aqueous HCl, then with brine, and dried with MgSO4. After the removal of the solvent, the residue was distilled in a vacuum. An effective deflegmator was necessary, due to difficulties in the separation of 1,4-bis(trimethylsilyl)buta-1,3-diyne that was formed as by-product. The yield was 3.0 g (77%); colorless oil, b.p. 118–120 °C (100 mbar). 1H-NMR (400 MHz, CDCl3) δ 0.26 (s, 9H, SiMe3), 8.35 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ-0.48 (s, SiMe3), 99.4 (s, C≡C), 100.2 (s, C≡C), 120.0 (q, 1JCF = 263.6 Hz, CF3), 135.4 (s, CPy), 137.2 (s, CPy), 144.5 (s, CPy), 151.8 (s, CPy). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 260 (100) [M]+. Anal. Calcd. for C10H11F3N2OSi: C, 46.14; H, 4.26; N, 10.76; Found: C, 46.19; H, 4.20; N, 10.69.
3.11. 2-Ethynyl-5-(trifluoromethoxy)pyrazine 17
The solution of 2-(trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16 (2 g, 7.7 mmol) and 18-crown-6 (0.05 g, 0.2 mmol) in THF (20 mL) was added dropwise at 5–10 °C to the solution of K2CO3 (1.1 g, 7.7 mmol) in 3 mL of water. The mixture was stirred for 2 h at 10–15 °C, neutralized with 5% aqueous HCl, and extracted with MTBE (5 × 25 mL). The organic extract was washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum. The yield was 1.2 g (85%); colorless oil or solid with a grass scent, b.p. 76–77 °C (10 mbar), m.p. 45–46 °C. 1H-NMR (400 MHz, CDCl3) δ 3.33 (s, 1H, ≡CH), 8.39 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 78.8 (s, ‒C≡), 81.4 (s, ≡CH), 119.9 (q, 1JCF = 263.6 Hz, CF3), 135.6 (s, CPy), 136.4 (s, CPy), 144.8 (s, CPy), 152.2 (s, CPy). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 188 (100) [M]+. Anal. Calcd. for C7H3F3N2O: C, 44.70; H, 1.61; N, 14.89; Found: C, 44.79; H, 1.60; N, 14.88.
3.12. 2-Ethynyl-5-methoxypyrazine 18
Method A. The solution of 2-(trifluoromethoxy)-5-((trimethylsilyl)ethynyl)pyrazine 16 (1 g, 3.8 mmol) in anhydrous methanol (3 mL) was added to 5 mL of the methanolic solution of KOH (0.43 g, 7.7 mmol) at 0 °C. The mixture was stirred for 2 h at 15 °C and the solvent was removed in a vacuum (without heating). The residue was suspended in ether and neutralized with 3% aqueous HCl. The organic solution was separated and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic solutions were washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum.
Method B. The solution of 2-Ethynyl-5-(trifluoromethoxy)pyrazine 17 (1 g, 5.3 mmol) in anhydrous methanol (3 mL) was added to 5 mL of the methanolic solution of KOH (0.43 g, 7.7 mmol) at −10 °C. The mixture was stirred for 2 h at 15 °C and the solvent was removed in a vacuum (without heating). The residue was suspended in ether and neutralized with 3% aqueous HCl. The organic solution was separated and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic solutions were washed with brine and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum.
The yield (Method A) was 0.34 g (65%), (Method B) 0.63 g (88%); colorless solid, m.p. 40–41 °C, b.p. 88–90 °C (10 mbar); 1H-NMR (400 MHz, CDCl3) δ 3.22 (s, 1H, ≡CH), 3.98 (s, 3H, CH3), 8.18 (s, 2H, 2 HPy), 8.25 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 53.9 (s, CH3), 78.9 (s, ≡CH), 80.0 (s, ‒C≡), 130.4 (s, CPy), 135.6 (s, CPy), 144.4 (s, CPy), 159.4 (s, CPy). GC-MS, 70 eV, m/z (rel. int.): 134 (100) [M]+. Anal. Calcd. for C7H6N2O: C, 62.68; H, 4.51; N, 20.88; Found: C, 62.62; H, 4.44; N, 20.80.
3.13. 1-(5-(Trifluoromethoxy)pyrazin-2-yl)ethan-1-one 19
Mercury oxide (0.38 g, 1.8 mmol) was added to the mixture of trifluoroacetic acid (60 mL, 800 mmol) and water (6 mL, 332 mmol) and stirred until dissolving (apr 10 min). The mixture was cooled to 10 °C and 2-ethynyl-5-(trifluoromethoxy)pyrazine 17 (1.0 g, 5.3 mmol) was added. The mixture was stirred at 40 °C for 1 h and trifluoroacetic acid was removed in a vacuum (300 mbar). Water (20 mL) was added to the residue and the mixture was neutralized with NaHCO3 to pH 6. The product was extracted with CHCl3 (3 × 10 mL) and the extract was washed with water and dried with MgSO4. After the removal of the solvent at atmospheric pressure, the residue was distilled in a vacuum. The yield was 0.92 g (84%); colorless solid, m.p. 44–45 °C, b.p. 94–95 °C (10 mbar); 1H-NMR (400 MHz, CDCl3) δ 2.69 (s, 3H, CH3), 8.42 (s, 2H, 2 HPy), 8.92 (s, 2H, 2 HPy). 13C-NMR (100 MHz, CDCl3) δ 25.9 (s, CH3), 119.9 (q, 1JCF = 264.6 Hz, CF3), 134.1 (s, CPy), 141.3 (s, CPy), 145.6 (s, CPy), 155.1 (s, CPy), 197.5 (s, CO). 19F-NMR (376.5 MHz, CDCl3) δ −57.3 (s, CF3). GC-MS, 70 eV, m/z (rel. int.): 206 (100) [M]+. Anal. Calcd. for C7H5F3N2O: C, 40.79; H, 2.45; N, 13.59; Found: C, 40.70; H, 2.44; N, 13.60.
4. Conclusions
It was shown that 2-chloro-5-trifluoromethoxypyrazine is suitable for further transformations via chlorine atom substitution in Buchwald-Hartwig amination, Kumada-Corriu, and Suzuki and Sonogashira coupling reactions. At the same time, the trifluoromethoxy group attached to the electron-deficient pyrazine ring was easily destructed and displaced under the action of O- or S- nucleophiles. This property must be taken into account, especially in the processes of bioactive molecules modeling. On the other hand, this fact opens up the possibility to explore trifluoromethoxy moiety as a leaving group for nucleophilic substitution.
Efficient and scalable methods for 2-chloro-5-trifluoromethoxypyrazine synthesis, developed in this work, and its synthetic profile allow us to consider this molecule as perspective for wide synthetic utility.
Supplementary Materials
1H-, 13C-, and 19F-NMR spectra of products associated with this article are available online.
Author Contributions
T.M.S. and Y.L.Y. conceived the idea of the article, conceived and performed the experiments, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Leroux, F.R.; Manteau, B.; Vors, J.-P.; Pazenok, S. Trifluoromethyl ethers—Synthesis and properties of an unusual substituent. Beilstein J. Org. Chem. 2008, 4. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Blakemore, D.C.; Narayanan, A.; Unwalla, R.; Lovering, F.; Denny, R.A.; Zhou, H.; Bunnage, M.E. Fluorine in drug design: A case study with fluoroanisoles. Chem. Med. Chem. 2015, 10, 715–726. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, K.N.; Ngai, M.-Y. Synthesis of tri- and difluoromethoxylated compounds by visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2019, 58, 11171–11181. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, W.A. The effect of fluorine substitution on the electronic properties of alkoxy, alkylthio and alkylsulfonyl groups. J. Am. Chem. Soc. 1963, 85, 1314–1318. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.-P.; Pazenok, S.; Langlois, B.R. A new and direct trifluoromethoxylation of fliphatic substrates with 2,4-dinitro(trifluoromethoxy)benzene. Adv. Synth. Catal. 2010, 35, 2831–2837. [Google Scholar] [CrossRef]
- Leo, A.; Jow, P.Y.C.; Silipo, C.; Hansch, C. Calculation of hydrophobic constant (log P) from π and f constants. J. Med. Chem. 1975, 18, 865–868. [Google Scholar] [CrossRef]
- Bzhezovskii, V.M.; Kapustin, E.G.; Yagupolskii, L.M. Stucture of C6H5XCF3 molecules (X=O or S): A quantum-chemical study. Russ. J. General. Chem. 2003, 73, 229–239. [Google Scholar] [CrossRef]
- Manteau, B.; Genix, P.; Brelot, L.; Vors, J.-P.; Pazenok, S.; Giornal, F.; Leuenberger, C.; Leroux, F.R. A general approach to (trifluoromethoxy)pyridines: First X-ray structure determinations and quantum chemistry studies. Eur. J. Org. Chem. 2010, 6043–6066. [Google Scholar] [CrossRef]
- Wang, L.; Wei, J.; Wu, R.; Cheng, G.; Li, X.; Hu, J.; Hu, Y.; Sheng, R. The stability and reactivity of tri-, di-, and monofluoromehtyl/methoxy/methylthio groups on arenes under acidic and basic conditions. Org. Chem. Front. 2017, 4, 214–223. [Google Scholar] [CrossRef]
- Yagupol’skii, L.M.; Troitskaya, V.I. Synthesis of derivatives of phenyl trifluoromethyl ether. Zhurnal Obschei Khimii. 1961, 31, 915–924. [Google Scholar]
- Olah, G.A.; Yamato, T.; Hashimoto, T.; Shih, J.G.; Trivedi, N.; Singh, B.P.; Piteau, M.; Olah, J.A. Electrophilic nitration, halogenation, acylation, and alkylation of α,α,α-trifluoromethoxy-benzene. J. Am. Chem. Soc. 1987, 109, 3708–3713. [Google Scholar] [CrossRef]
- Mandal, D.; Gupta, R.; Jaiswal, A.K.; Young, R.D. Frustrated Lewis pair meditated selective single fluoride substitution in trifluoromethyl groups. Am. Chem. Soc. 2020, 142, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; John Wiley & Sons Ltd.: Chichester, UK, 2010; 718p. [Google Scholar]
- Maga, J.A.; Katz, I. Pyrazines in foods: An update. Food Sci. Nutr. 1982, 16, 1–48. [Google Scholar] [CrossRef]
- Osada, K.; Miyazono, S.; Kashiwayanagi, M. The scent of wolves: Pyrazine analogs induce avoidance and vigilance behaviors in prey. Front. Neurosci. 2015, 9, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, V.J.; Mahendra, A.S.; Pratap, N.R. The Chemistry of Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019; 503p. [Google Scholar]
- Yagupolskii, L.M. Synthesis of derivatives of phenyl trifluoromethyl ethers. Dokl. Acad. Nauk SSSR 1955, 105, 100–102. [Google Scholar]
- Kanie, K.; Tanaka, Y.; Shimizu, M.; Kuroboshi, M.; Hiyama, T. Oxidative desulfurization-fluorination ofalkanol xanthates. Control of the reaction pathway to fluorination or trifluoromethoxylation. Chem. Commun. 1997, 309–310. [Google Scholar] [CrossRef]
- Ben-David, I.; Rechavi, D.; Mishani, E.; Rozen, S. A novel synthesis of trifluoromethyl ethers via xanthates utilizing BrF3. J. Fluor. Chem. 1999, 97, 75–78. [Google Scholar] [CrossRef]
- Blazejewski, J.C.; Anselmi, E.; Wakselman, C. 2-Trifluoromethoxyethyl triflate: A versatile trifluoromethoxyethyl carrier. J. Org. Chem. 2001, 66, 1061–1063. [Google Scholar] [CrossRef]
- Sokolenko, T.M.; Dronkina, M.I.; Magnier, E.; Yagupolskii, L.M.; Yagupolskii, Y.L. Evaluation of efficient and practical methods for the preparation of functionalized aliphatic trifluoromethyl ethers. Molecules 2017, 22, 804. [Google Scholar] [CrossRef] [Green Version]
- Yoritate, M.; Londregan, A.T.; Lian, Y.; Hartwig, J.F. Sequential xanthalation and O-trifluoromethylation of phenols: A procedure for the synthesis of aryl trifluoromethyl ethers. J. Org. Chem. 2019, 84, 15767–15776. [Google Scholar] [CrossRef]
- Umemoto, T.; Adachi, K.; Ishihara, S. CF3 Oxonium salts, O-(trifluoromethyl)dibenzofuranium salts: In situ synthesis, properties, and application as a real CF3 species reagent. J. Org. Chem. 2007, 72, 6905–6917. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, J.; Fruh, N.; Togni, A. Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 2015, 115, 650–682. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.; Han, S.; Liu, Z.; Wang, L.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Regioselective synthesis of N-heteroaromatic trifluoromethoxy compounds by direct O‒CF3 bond formation. Chem. Eur. J. 2016, 22, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Song, X.; Guo, J.; Chen, S.; Gao, J.; Jiang, J.; Gao, F.; Li, Y.; Wang, M. Synthesis of chloro(phenyl)trifluoromethyliodane and catalyst-free electrophilic trifluoromethylations. Org. Lett. 2018, 20, 3933–3937. [Google Scholar] [CrossRef] [PubMed]
- Liebing, P.; Kalim, J.; Arefyeva, N.; Oehler, F.; Wickleder, M.; Togni, A. A tunable trifluoromethyliodonium reagent. Angew. Chem. Int. Ed. 2019, 58, 8585–8588. [Google Scholar] [CrossRef]
- Kalim, J.; Duhail, T.; Le, T.-N.; Vanthuyne, N.; Anselmi, E.; Togni, A.; Magnier, E. Merging hypervalent Iodine and sulfoximine chemistry: A new electrophilic trifluoromethylation reagent. Chem. Sci. 2019, 45, 10516–10523. [Google Scholar] [CrossRef]
- Farnham, W.B.; Middleton, W.J. Tris(disubstituted amino)sulfonium Perfluoroalkoxides and Perfluoroalkylmercaptides and Process for Their Preparation. U.S. Patent 4628094, 9 December 1986. [Google Scholar]
- Kolomeitsev, A.A.; Vorobyev, M.; Gillandt, H. Versatile application of trifluoromethyl triflate. Tetrahedron Lett. 2008, 49, 449–454. [Google Scholar] [CrossRef]
- Koller, R.; Huchet, Q.; Battaglia, P.; Welch, J.M.; Togni, A. Acid-mediated formation of trifluoromethyl sulfonates from sulfonic acids and a hypervalent iodine trifluoromethylating agent. Chem. Commun. 2009, 5993–5995. [Google Scholar] [CrossRef]
- Sokolenko, T.M.; Davydova, Y.A.; Yagupolskii, Y.L. Efficient synthesis of 5′-fluoroalkoxythiazoles via α-bromo-α-fluoroalkoxyacetophenones Hantzsch type cyclization with thioureas or thioamides. J. Fluorine Chem. 2012, 136, 20–25. [Google Scholar] [CrossRef]
- Davydova, Y.A.; Sokolenko, T.M.; Yagupolskii, Y.L. Polyfluoro- and perfluoroalkoxyenaminones in syntheses of nitrogen containing heterocycles. J. Fluorine Chem. 2014, 157, 58–62. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, P. Recent advances in new trifluoromethoxylation reagents. Sci. China Chem. 2019, 62. [Google Scholar] [CrossRef]
- Nikishkin, N.I.; Huskens, J.; Verboom, W. Transition metal-catalyzed functionalization of pyrazines. Org. Biomol. Chem. 2013, 11, 3583–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Ogata, T.; Hartwig, J.F. Highly reactive, general and long-lived catalysts for palladium-catalyzed amination of heteroaryl and aryl chlorides, bromides, and iodides: Scope and structure–activity relationships. J. Am. Chem. Soc. 2008, 130, 6586–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratenko, N.V.; Kolomeitsev, A.A.; Mogilevskaya, V.O.; Varlamova, N.M.; Yagupolskii, L.M. Polynitro- and poly[(trifluoromethyl)sulfonyl]substituted diphenylamines. Zhurn. Org. Khimii 1986, 22, 1721–1729. [Google Scholar]
- Seifert, R.M.; Buttery, R.G.; Guadagni, D.G.; Black, D.R.; Harris, J.G. Synthesis of some 2-methoxy-3-alkylpyrazines with strong bell pepper-like odors. J. Agric. Food Chem. 1970, 18, 246–249. [Google Scholar] [CrossRef]
- Seifert, R.M.; Buttery, R.G.; Guadagni, D.G.; Black, D.R.; Harris, J.G. Synthesis and odor properties of some additional compounds related to 2-isobutyl-3-methoxypyrazine. J. Agric. Food Chem. 1972, 20, 135–137. [Google Scholar] [CrossRef]
- Shibamoto, T. Odor threshold of some pyrazines. J. Food Sci. 1986, 51, 1098–1099. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Masse, J.P. Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls. J. Chem. Soc. Chem. Commun. 1972, 144. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. Iron-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Sonogashira, K. Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kondo, Y.; Shiraiwa, M.; Yamanaka, H. A facile synthesis of ethynyl-substituted six-membered N-heteroaromatic compounds. Synthesis 1983, 312–314. [Google Scholar] [CrossRef]
- Hassner, A.; Stumer, C. Organic Syntheses Based on Name Reactions and Unnamed Reactions; Pergamon: Kidlington, UK, 1994; p. 219. [Google Scholar]
- Sakamoto, T.; Kondo, Y.; Shiraiwa, M.; Yamanaka, H. ω-Methoxylation and hydration of ethynyl-N-heteroarenes. Synthesis 1984, 245–247. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals, 8th ed.; Butterworth-Heinemann: Oxford, UK, 2008; p. 1198. [Google Scholar]
- Palamidessi, G.; Bernardi, L. On 2,5-dichloropyrazine. J. Org. Chem. 1964, 29, 2491–2492. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–19 are available from the authors. |
Scheme 1.
Preparation of the 2-chloro-5-trifluoromethoxypyrazine.

Scheme 2.
Reaction of 2-chloro-5-trifluoromethoxypyrazine with 3-methylthiophenol.

Scheme 3.
Probable mechanism of OCF3 group destruction with the S-nucleophiles.

Scheme 4.
2-Chloro-5-trifluoromethoxypyrazine 4 in the Buchwald-Hartwig amination reaction.

Scheme 5.
2-Chloro-5-trifluoromethoxypyrazine 4 in the palladium-catalyzed cyanation reaction.

Scheme 6.
2-Chloro-5-trifluoromethoxypyrazine 4 in the iron-catalyzed Kumada–Corriu coupling.

Scheme 7.
2-Chloro-5-trifluoromethoxypyrazine 4 in Suzuki coupling.

Scheme 8.
2-Chloro-5-trifluoromethoxypyrazine Sonogashira coupling and Kucherov hydration.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sokolenko, T.M.; Yagupolskii, Y.L. Trifluoromethoxypyrazines: Preparation and Properties. Molecules 2020, 25, 2226. https://doi.org/10.3390/molecules25092226
AMA Style
Sokolenko TM, Yagupolskii YL. Trifluoromethoxypyrazines: Preparation and Properties. Molecules. 2020; 25(9):2226. https://doi.org/10.3390/molecules25092226
Chicago/Turabian StyleSokolenko, Taras M., and Yurii L. Yagupolskii. 2020. "Trifluoromethoxypyrazines: Preparation and Properties" Molecules 25, no. 9: 2226. https://doi.org/10.3390/molecules25092226