* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
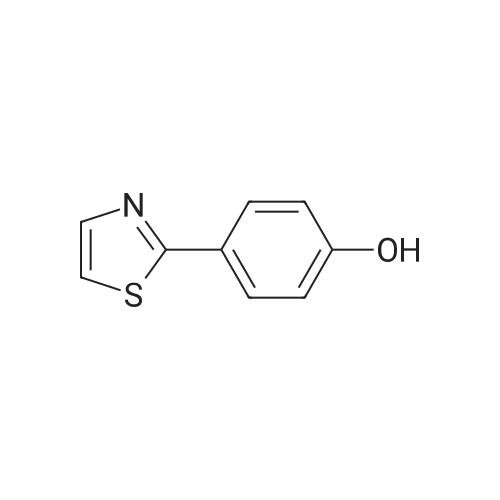
[1] Journal of Medicinal Chemistry, 1986, vol. 29, # 6, p. 1065 - 1080
2
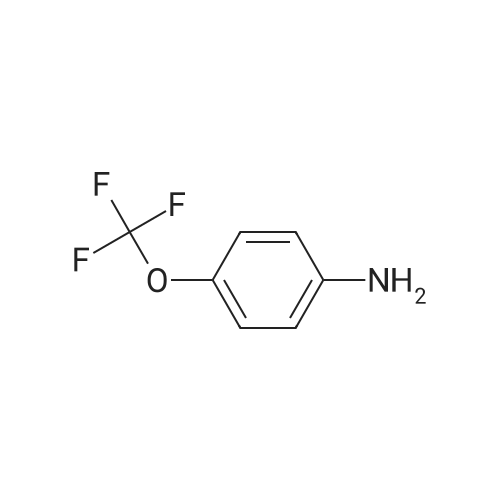
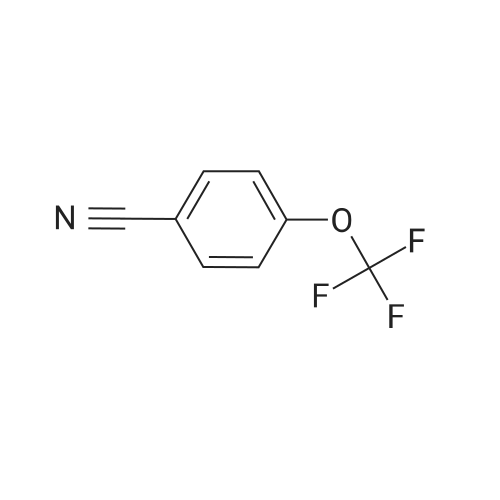
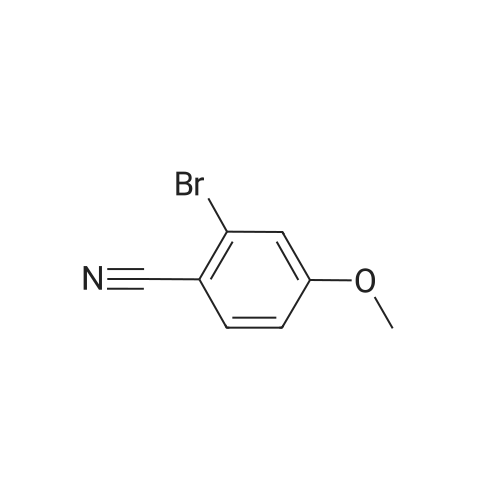
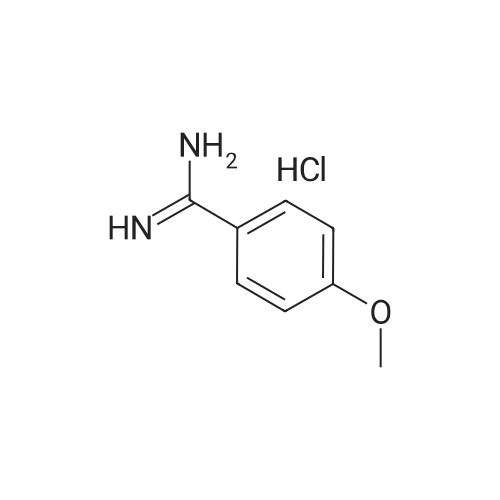
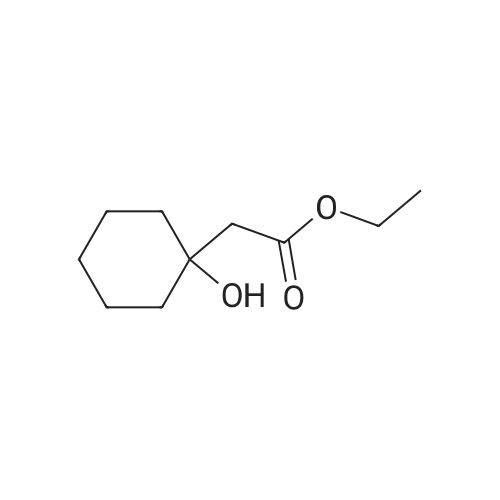
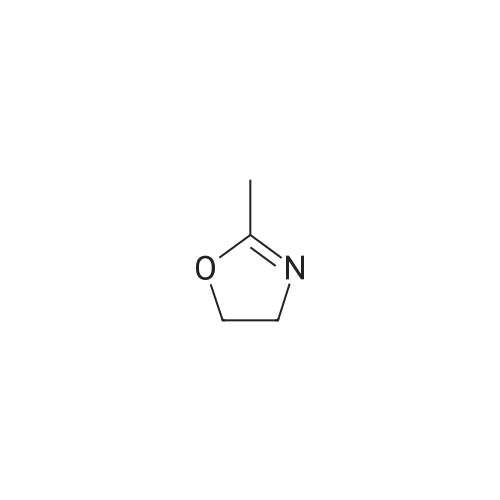
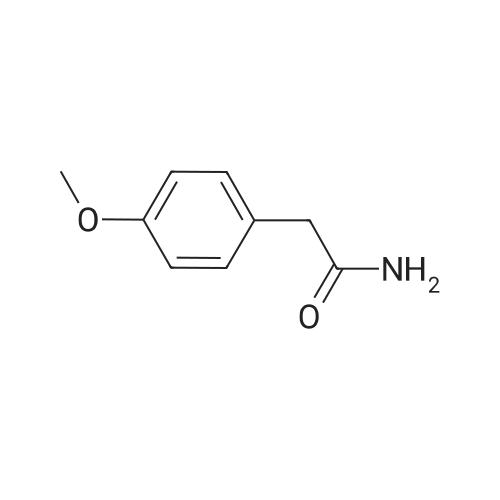
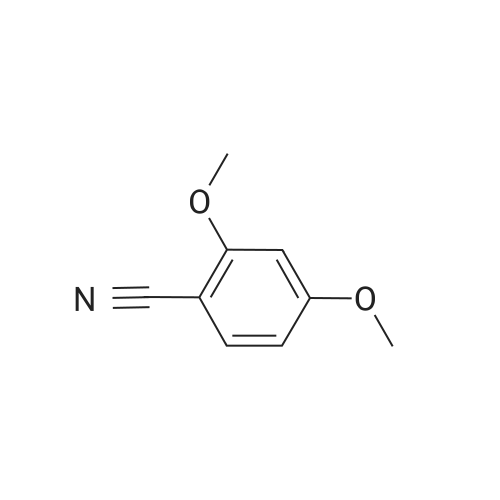
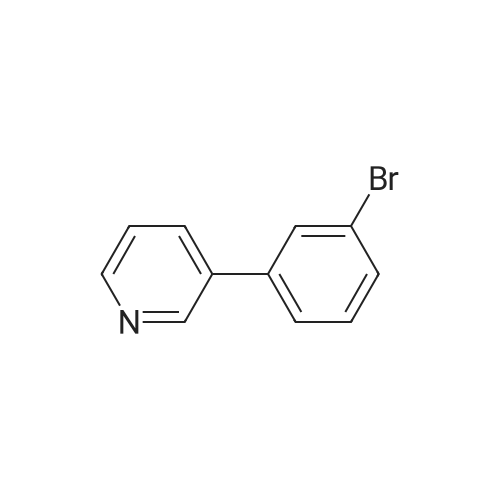
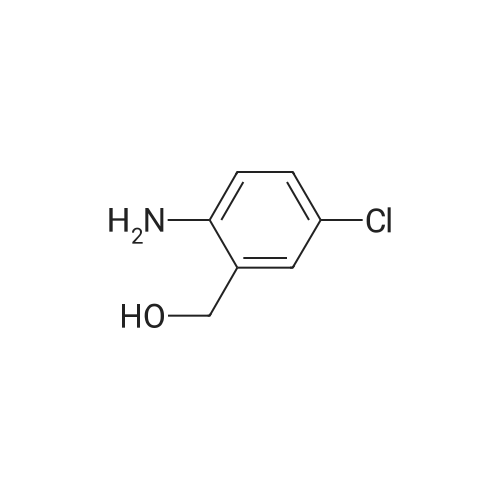
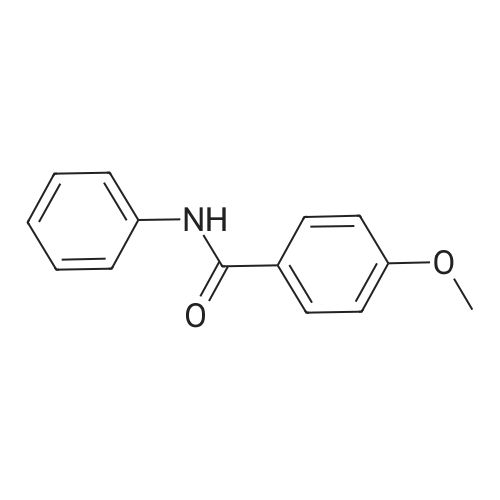
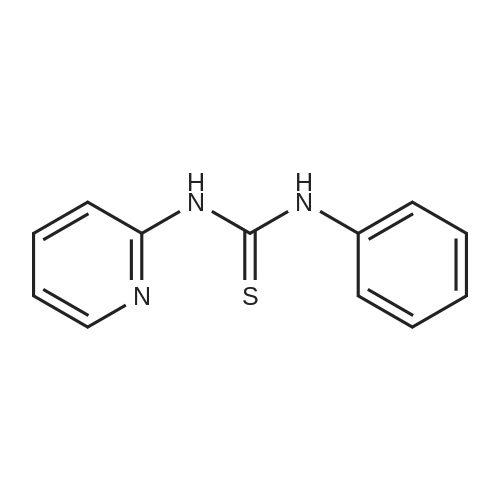
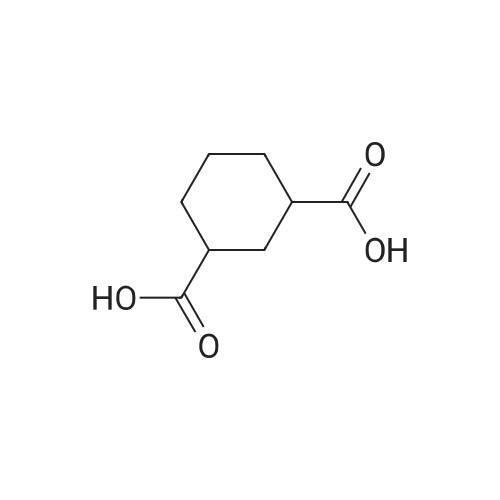
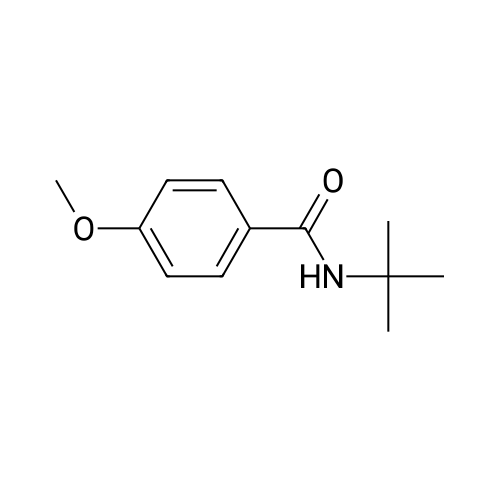
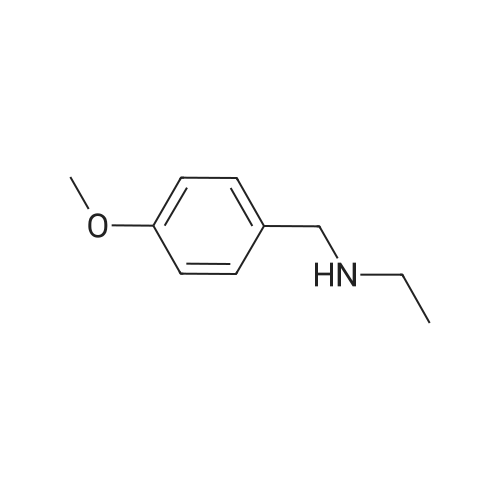
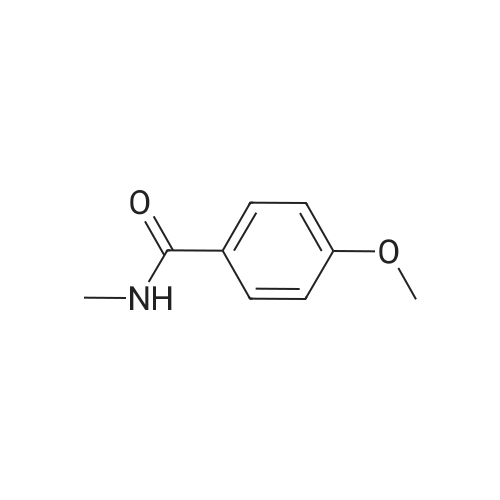
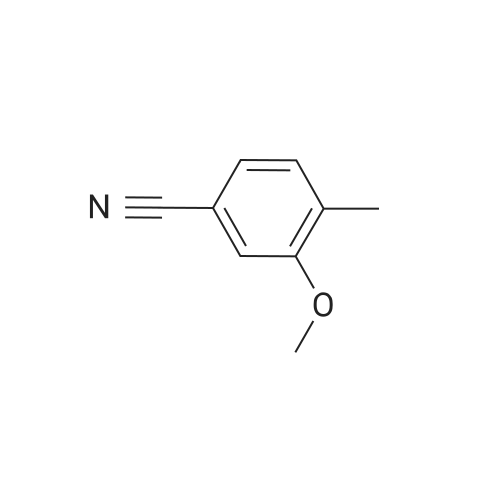
[ 79-19-6 ]
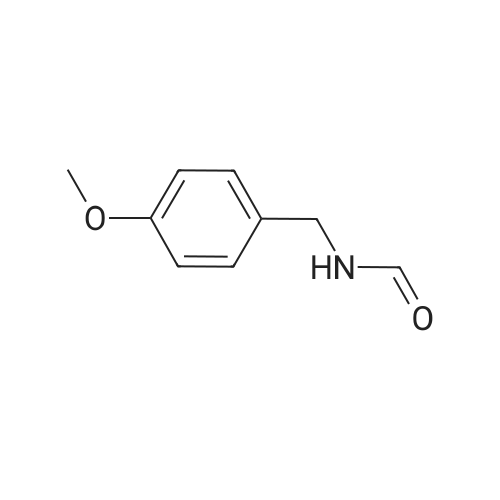
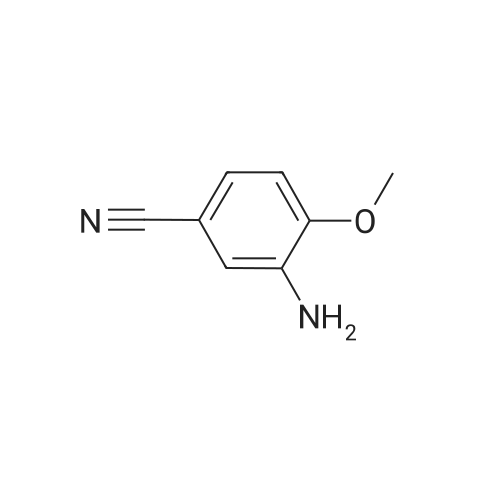
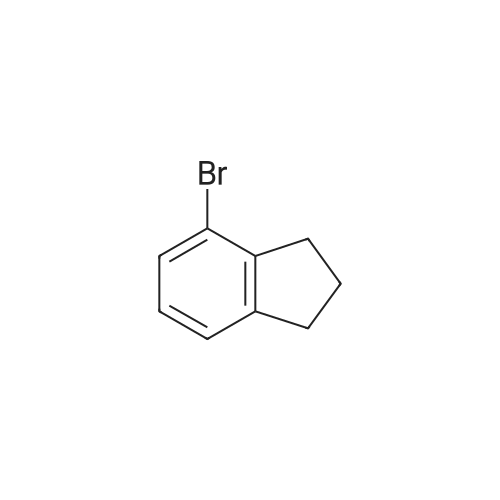
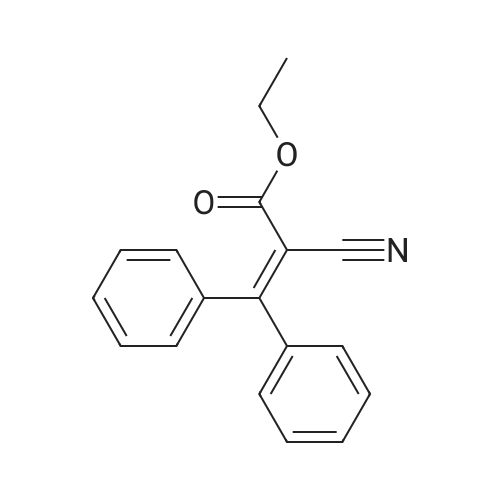
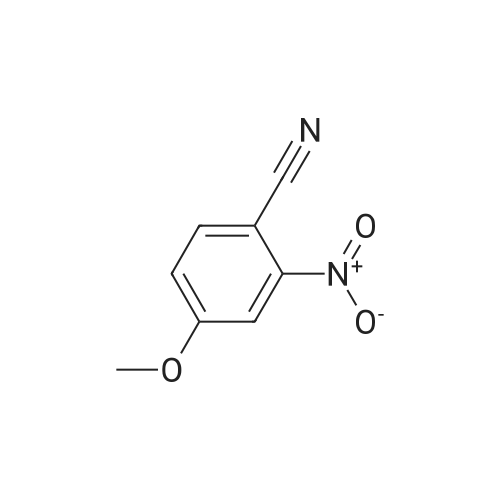
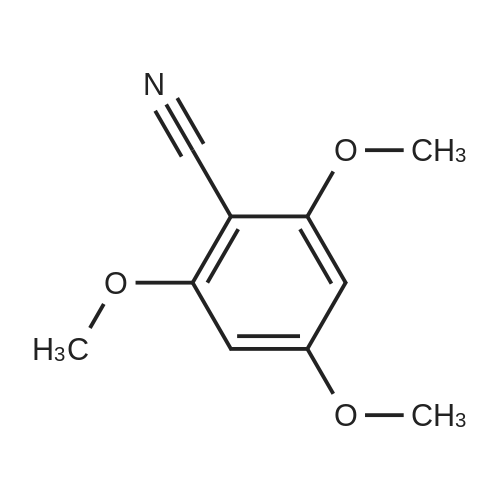
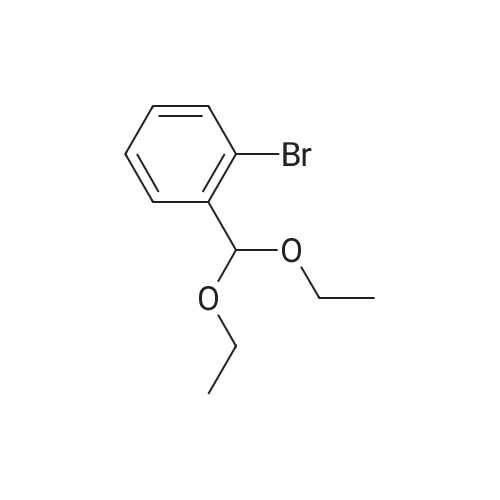
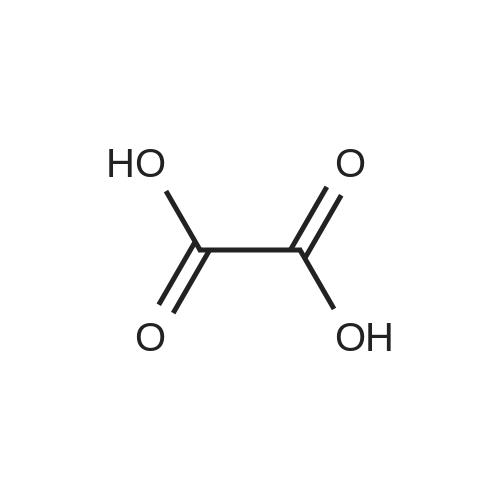
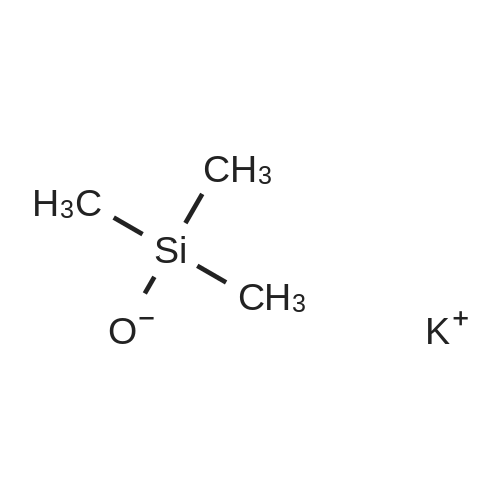
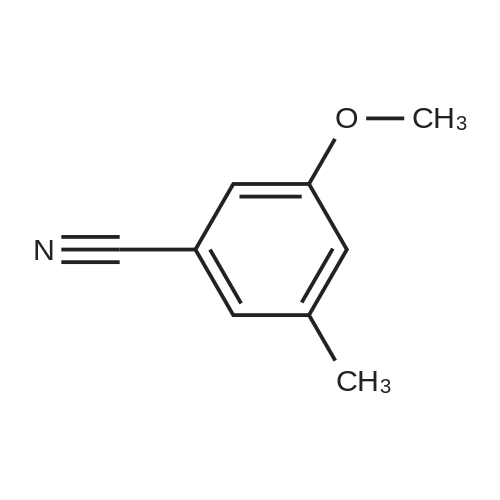
[ 874-90-8 ]
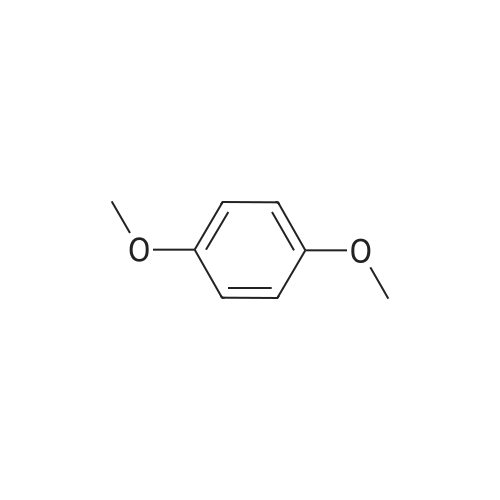
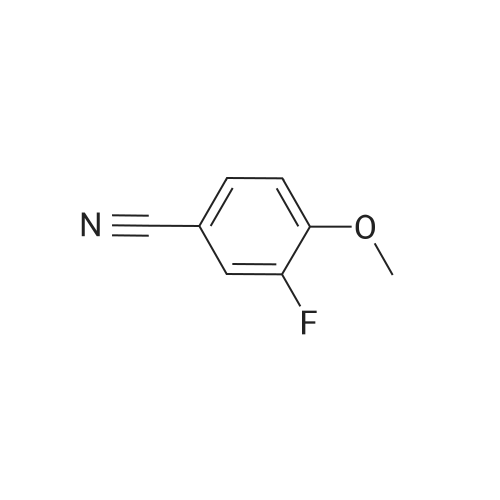
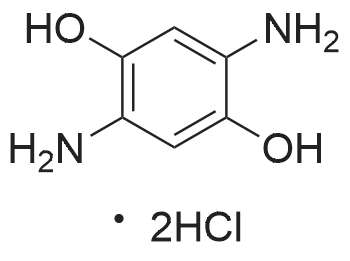
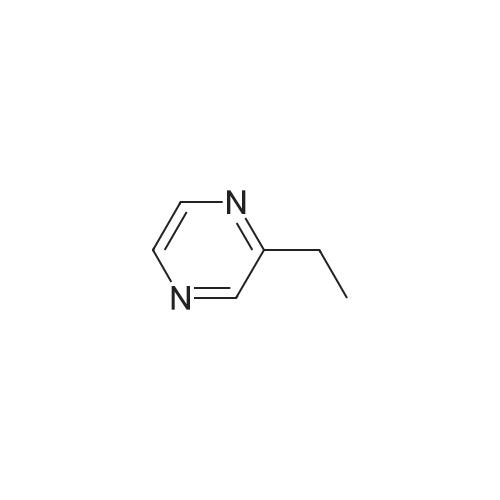
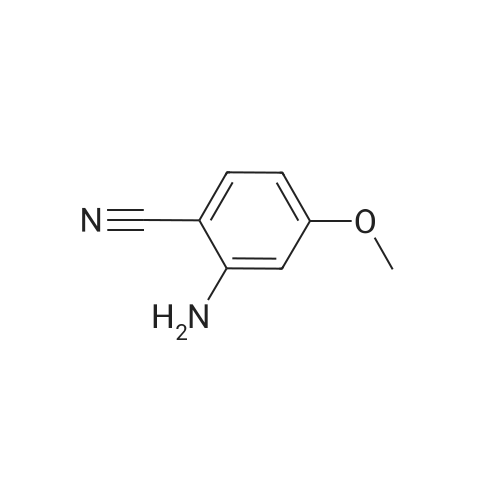
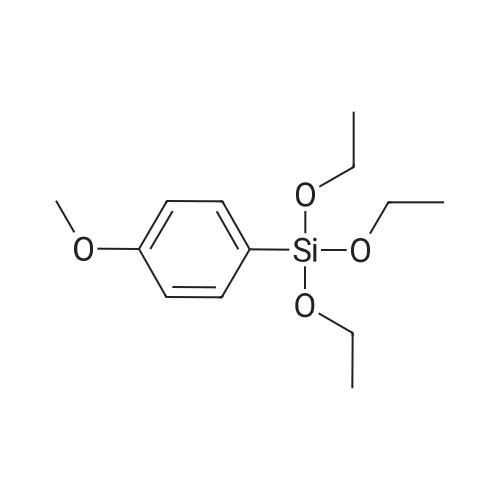
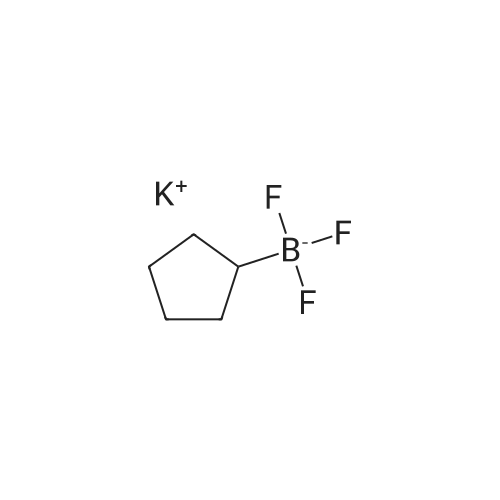
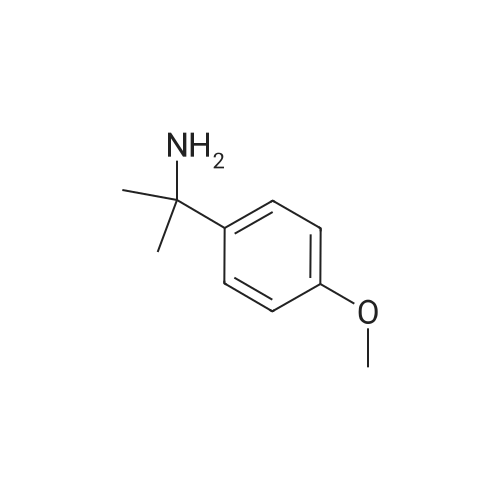
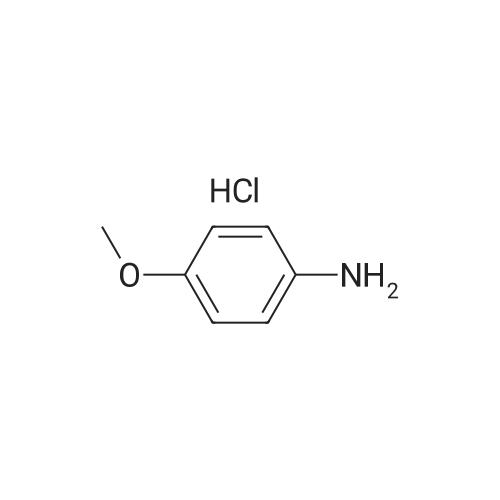
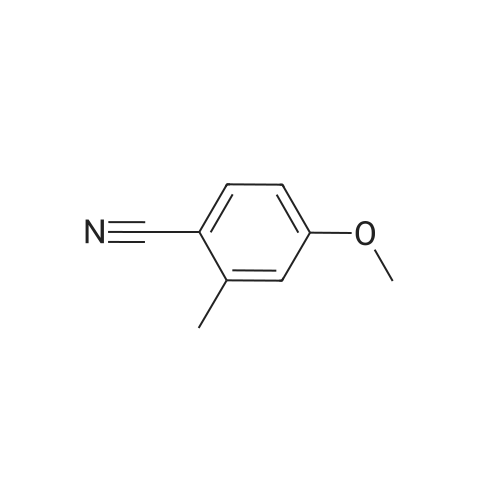
[ 1014-25-1 ]
Reference:
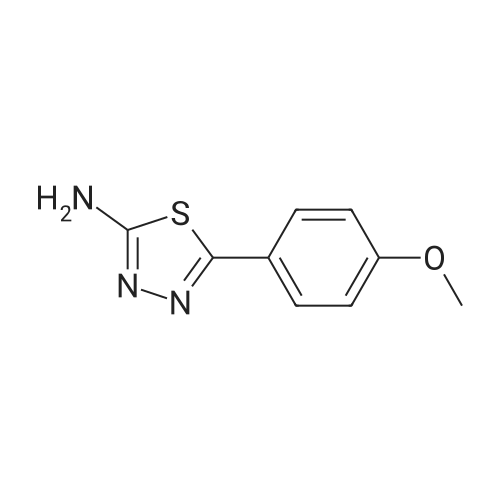
[1] Russian Chemical Bulletin, 1995, vol. 44, # 10, p. 1955 - 1956[2] Izvestiya Akademi Nauk, Seriya Khimicheskaya, 1995, # 10, p. 2035 - 2036
3
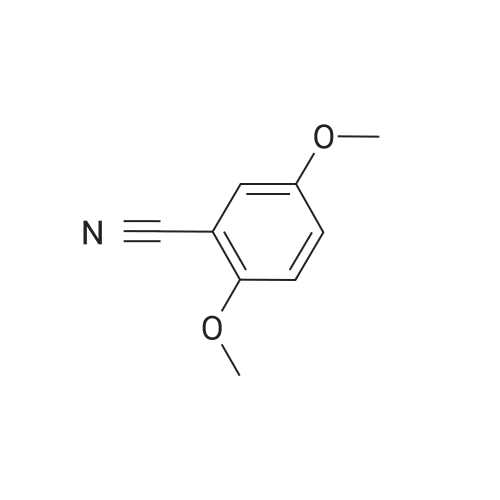
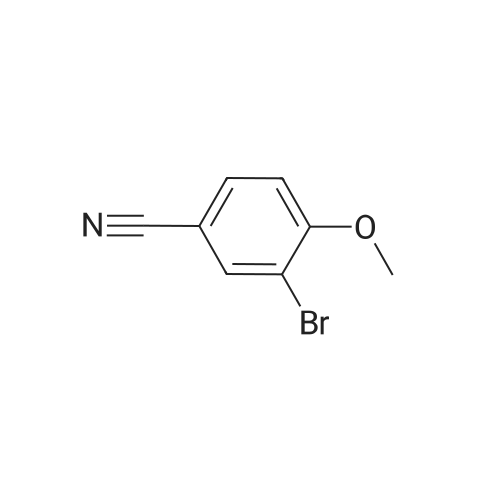
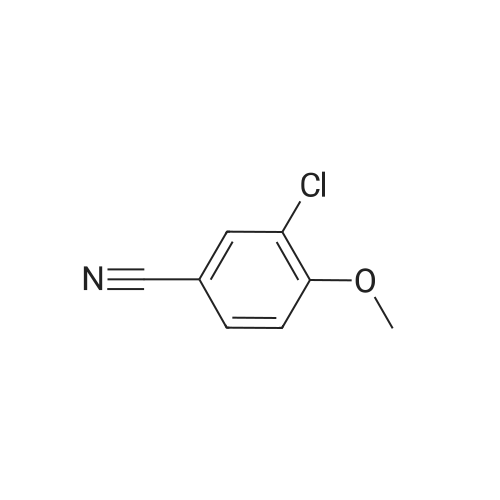
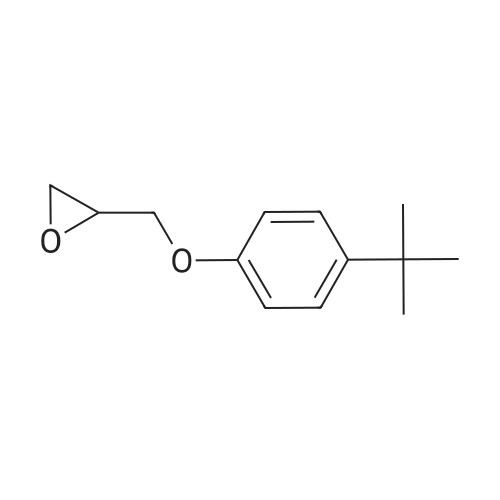
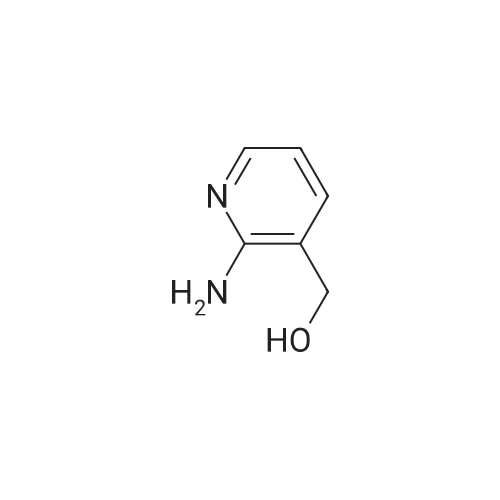
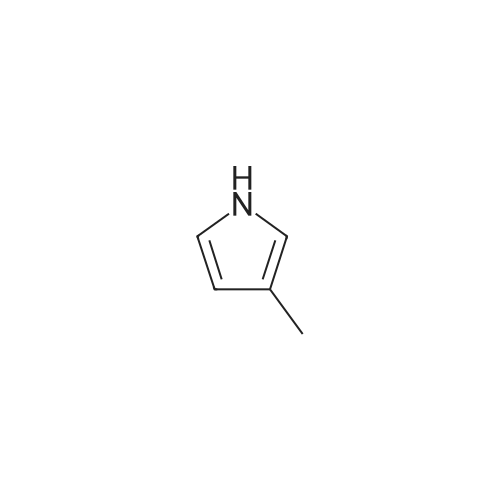
[ 78514-88-2 ]
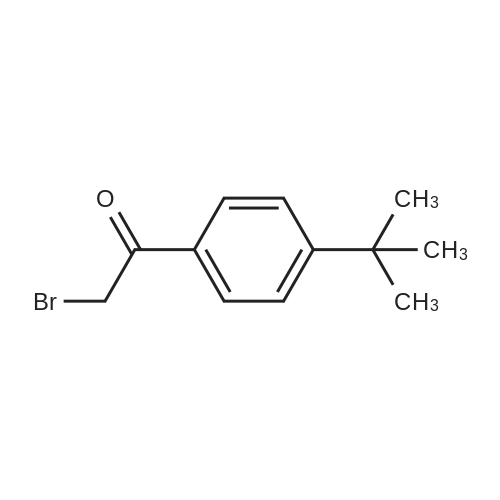
[ 776-53-4 ]
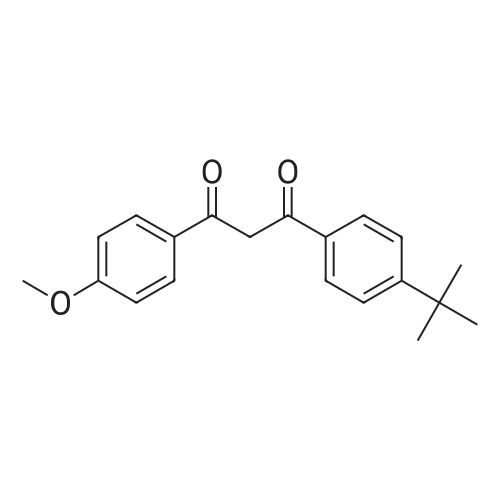
[ 78515-01-2 ]
[ 874-90-8 ]
Reference:
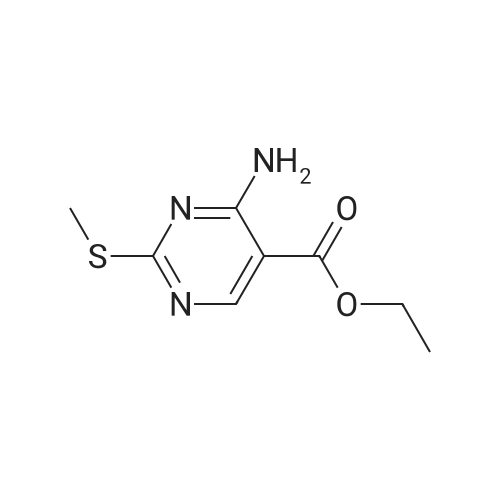
[1] Journal of Organic Chemistry, 1981, vol. 46, # 20, p. 3956 - 3959
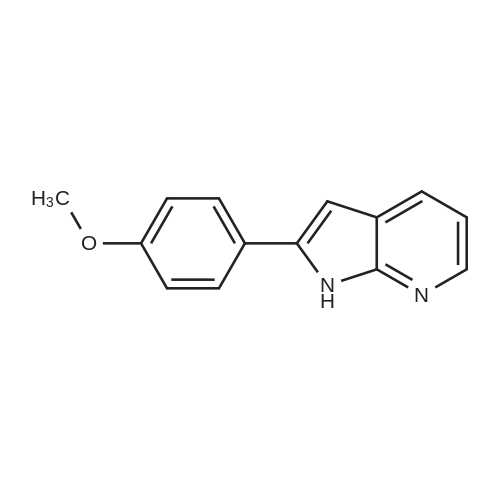
4
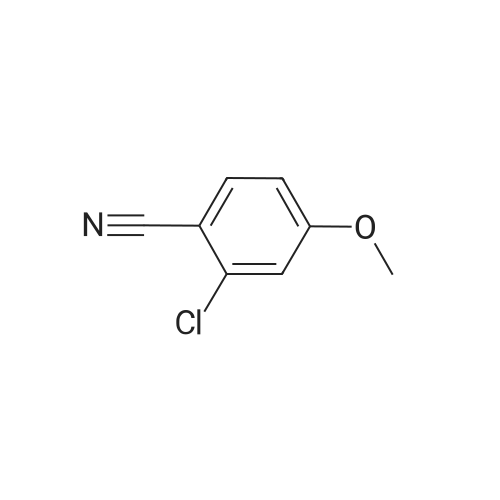
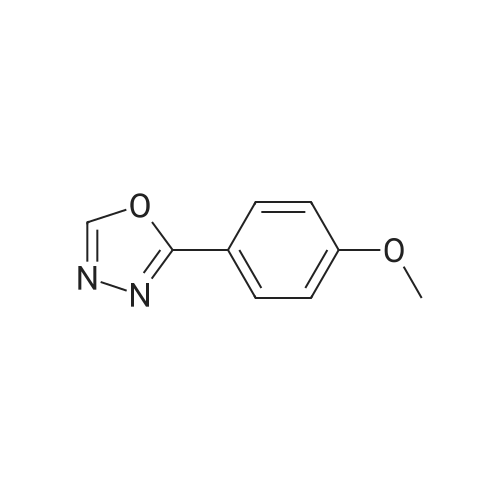
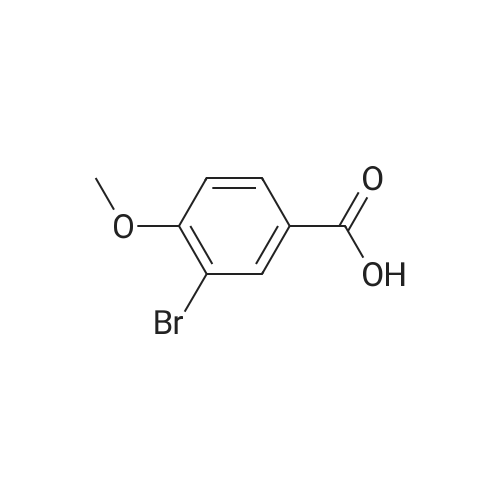
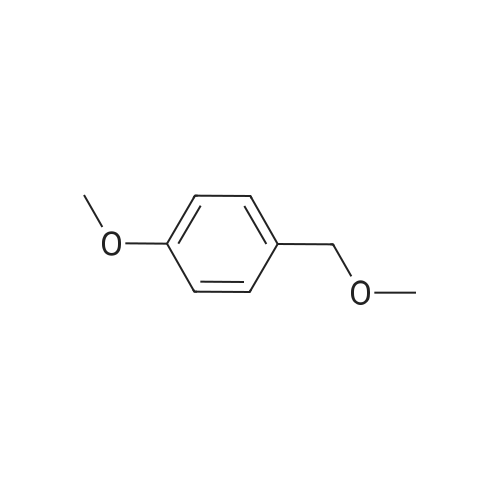
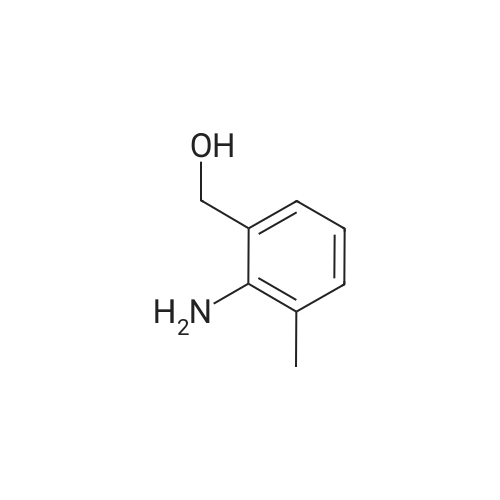
[ 874-90-8 ]
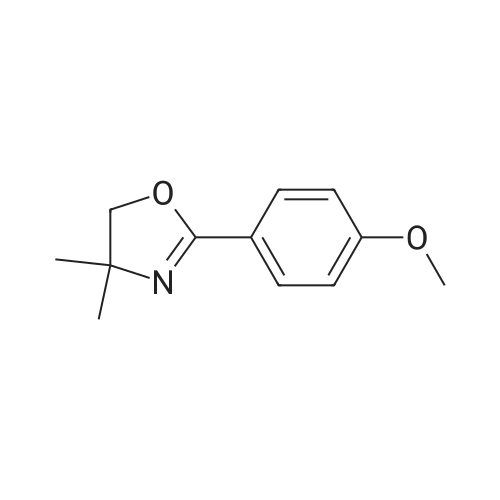
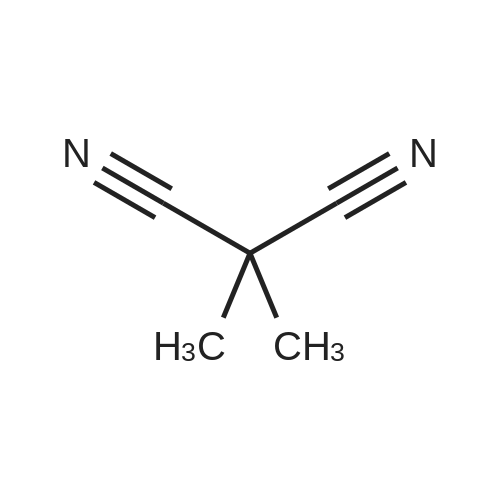
[ 2881-83-6 ]
Yield
Reaction Conditions
Operation in experiment
94%
Stage #1: at 0 - 25℃; for 92 h; Stage #2: With hydrogenchloride In tetrahydrofuran; water at 20 - 25℃; for 1.58333 h;
Under nitrogen atmosphere, 30 ML of THF was added to 6.09 g (10 mmol, 1.0 equivalent) of (BrZnCH2COOEt*THF)2. Under argon atmosphere, a solution of 1.33 g (10 mmol) of anisonitrile in 5 ML of THF was added dropwise while stirring at 0.similar.5°C. The mixture was stirred at 20.similar.25°C for 92 hours. 15 ML of 10percent hydrochloric acid was added dropwise at 20°C or lower, and the mixture was stirred at 20.similar.25°C for 1 hour and 35 minutes, followed by dilution with 50 ML of ethyl acetate.. Then, the layers were separated.. The organic layer was washed successively with 15 ML of 1N hydrochloric acid, 20 ML of an aqueous saturated sodium chloride solution, 20 ML (*2) of an aqueous saturated sodium bicarbonate solution, and 20 ML of an aqueous saturated sodium chloride solution.. After washing, the organic layer was dried with anhydrous magnesium sulfate.. Concentration under reduced pressure afforded 2.08 g of the desired product (yield 94percent).1H NMR (CDCl3), (ppm): δ [1.25 (t, J=7.1 Hz), 1.33 (t,J=7.1 Hz)] (3H), 3.87 (3H, s), [3.94 (s), 5.58 (s), 12.6 (s)] (2H), 4.17-4.24 (2H, m), 6.94 (d, 2H, J=8.8 Hz), 7.93 (d, 2H, J=8.8 Hz).
Reference:
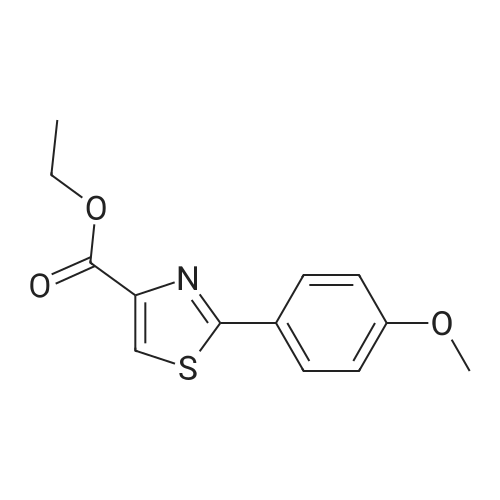
[1] Journal of Organic Chemistry, 2013, vol. 78, # 6, p. 2786 - 2791
19
[ 874-90-8 ]
[ 51721-68-7 ]
Yield
Reaction Conditions
Operation in experiment
44.5%
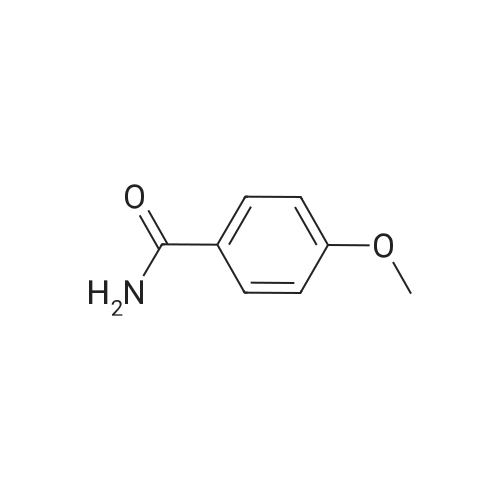
Stage #1: With sodium methylate In methanol at 20℃; for 48 h; Stage #2: With ammonium chloride In methanol at 20℃; for 24 h;
A solution of 4-methoxybenzonitrile 2 (2.1 g, 15.7 mmol), sodium methylate (86 mg, 1.57 mmol) in 20 mL of anhydrous methanol was stirred at room temperature for 48 h. Then ammonium chloride (0.84 g, 15.7 mmol) was added, and the reaction group was stirred for another 24 h the precipitate was collected by filtration, washed with diethyl ether, and then dried in vacuum to obtain 3 as a white solid (1.3 g, 44.5percent), MS (ESI) m/z: 151.1 ([MH]).
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 150, p. 783 - 795
[2] Journal of the American Chemical Society, 1985, vol. 107, # 9, p. 2743 - 2748
[3] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 3, p. 613 - 623
[4] Collection of Czechoslovak Chemical Communications, 1981, vol. 46, # 4, p. 933 - 940
[5] Patent: US6218538, 2001, B1,
[6] European Journal of Organic Chemistry, 2014, vol. 2014, # 17, p. 3614 - 3621
[7] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 29 - 43
[8] Tetrahedron Letters, 2018, vol. 59, # 4, p. 361 - 364
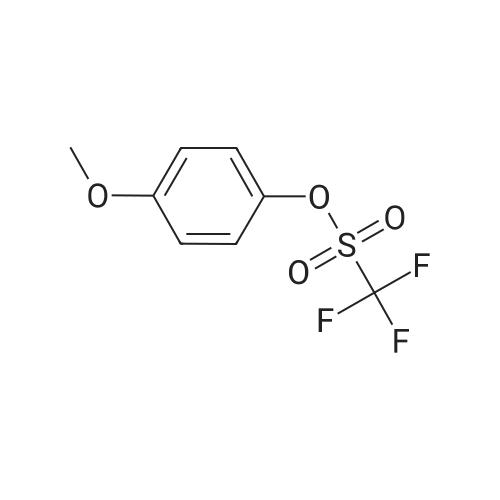
20
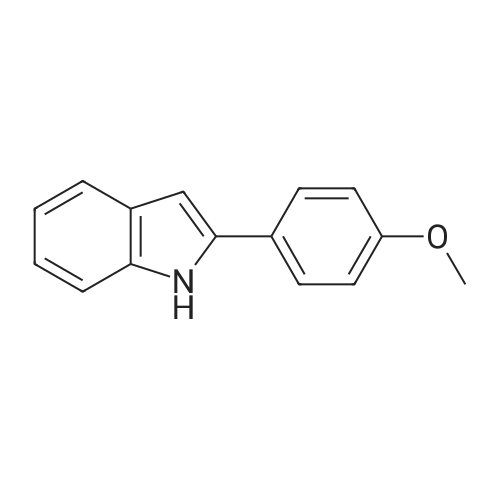
[ 874-90-8 ]
[ 102151-33-7 ]
Reference:
[1] Chemical Science, 2017, vol. 8, # 10, p. 7009 - 7013
21
[ 874-90-8 ]
[ 30095-47-7 ]
[ 70356-09-1 ]
Reference:
[1] European Journal of Organic Chemistry, 2015, vol. 2015, # 7, p. 1525 - 1532
22
[ 122836-11-7 ]
[ 15529-49-4 ]
[ 874-90-8 ]
[ 2393-23-9 ]
Reference:
[1] Journal of molecular catalysis, 1989, vol. 50, # 3, p. 333 - 341
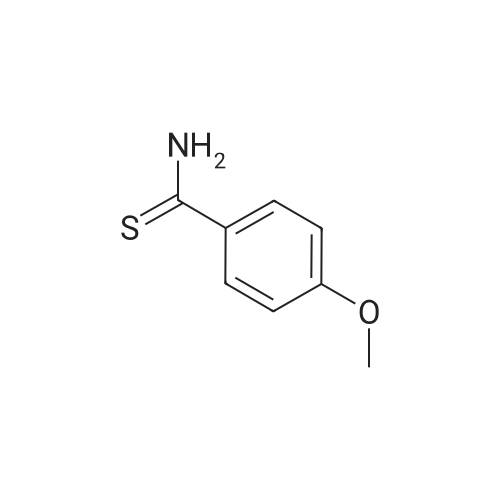
Stage #1: 4-methoxybenzonitrile With Lawessons reagent; boron trifluoride diethyl etherate In diethyl ether; toluene at 50℃; for 14h; Inert atmosphere;
Stage #2: With sodium hydrogencarbonate In diethyl ether; toluene at 20℃; for 0.333333h; Inert atmosphere;
93%
With aluminium trichloride; tiolacetic acid In 1,2-dichloro-ethane Ambient temperature;
93%
With sodium hydrogensulfide; magnesium chloride In N,N-dimethyl-formamide at 20℃; for 4h;
90%
With magnesium(II) chloride hexahydrate; sodium hydrogen sulfide monohydrate In N,N-dimethyl-formamide at 20℃; for 8h;
89%
With diammonium sulfide; 1,6-bis(3-methylimidazolium-1-yl)hexane dichloride at 70℃; for 0.833333h;
81%
With diphenylphosphinodithioic acid In isopropyl alcohol at 40 - 60℃;
80%
With Dowex 1X8 SH(1-); hydrogen sulfide In methanol at 20℃; for 6h;
79.6%
With pyridine; diammonium sulfide; triethylamine In water at 50℃;
76%
With 2,3,4,5,7,8,9,10-octahydropyrimido[1,2-a]azepin-1-ium acetate; sodiumsulfide nonahydrate In neat (no solvent) at 20℃; for 2h; Green chemistry;
73%
With hexamethyldisilathiane; sodium methylate In various solvent(s) at 35℃; for 24h;
70%
With sodiumsulfide nonahydrate In N,N-dimethyl-formamide at 130℃; for 2.5h;
The general procedure of preparing all thioamides
General procedure: Benzonitrile 1a (1 mmol), Na2S*9H2O (1.2 mmol) and DMF (1 mL) were added into a 10 mL bottle. The reactor was placed in a heating magnetic stirrer at 130 °C. After 2.5 h, by adding about 3 mL H2O after the reaction to disperse the solid product, the reaction mixture was extracted with EtOAc (3 x 3 mL), and the mixture was purified by column chromatography.
With diphenylphosphinodithioic acid In isopropyl alcohol
With hydrogen sulfide; triethylamine In pyridine at 18 - 22℃; effect of mole fraction triethylamine on rate constants;
With ammonium sulfide at 100℃; im Druckrohr;
With pyridine; hydrogen sulfide; triethylamine
With hydrogen sulfide; triethylamine
With pyridine; hydrogen sulfide; triethylamine Ambient temperature;
With hydrogen sulfide
With triethanolamine; hydrogen sulfide In ethanol
With hydrogenchloride; thioacetamide In N,N-dimethyl-formamide at 100℃; for 2h;
With pyridine; hydrogen sulfide; triethylamine
With O,O-Diethyl hydrogen phosphorodithioate In tetrahydrofuran at 66℃; for 5h; Heating / reflux;
1; 5
Example 1; Reactivity of Decoy Agents; One mmol of the aromatic nitrile decoy agents set forth in Table 1 were reacted at reflux with the thionating agent diethyl dithiophosphate (0.2 mL) in wet THF (6 mL) to give the respective thioamides. TABLE 1 Nitrile % Conversion to Thioamide* 2-thiophenecarbonitrile 100 benzonitrile 55 p-chlorobenzonitrile 67 p-methoxybenzonitrile 45 o-nitrobenzonitrile 25 p-acetylbenzonitrile 57 p-methylbenzonitrile 52 p-fluorobenzonitrile 68 * % conversion as determined by gas chromatography/mass spectroscopy (GC/MS) This example illustrates that 2-thiophenecarbonitrile was the most reactive with the thionating agent.; Example 5; Effect of Acetonitrile on the Formation of Thioamide Impurities; The nitrile set forth in Table 4 was reacted at reflux with diethyl dithiophosphate (200 μL) in wet THF (5 mL) and acetonitrile (1 mL=20 molar equivalents). The control set contained 6 mL THF and no acetonitrile. After 5 hours at 66° C., the mixtures were subjected to GC/MS analysis to detect the presence of thioamide impurity. TABLE 4 % Conversion to Thioamide Nitrile with acetonitrile without acetonitrile 2-thiophenecarbonitrile 33 78 benzonitrile 11 43 p-acetylbenzonitrile 11 45 p-methoxybenzonitrile 2 38 p-chlorobenzonitrile 13 51 1,4-dicyanobenzene 34 67 This example illustrates that conversion to the thioamide impurity was suppressed in samples containing acetonitrile. Further, samples containing acetonitrile and p-methoxybenzonitrile had very little conversion to the thioamide impurity.
With O,O-Diethyl hydrogen phosphorodithioate; acetonitrile In tetrahydrofuran at 66℃; for 5h;
5
Example 5; Effect of Acetonitrile on the Formation of Thioamide Impurities; The nitrile set forth in Table 4 was reacted at reflux with diethyl dithiophosphate (200 μL) in wet THF (5 mL) and acetonitrile (1 mL=20 molar equivalents). The control set contained 6 mL THF and no acetonitrile. After 5 hours at 66° C., the mixtures were subjected to GC/MS analysis to detect the presence of thioamide impurity. TABLE 4 % Conversion to Thioamide Nitrile with acetonitrile without acetonitrile 2-thiophenecarbonitrile 33 78 benzonitrile 11 43 p-acetylbenzonitrile 11 45 p-methoxybenzonitrile 2 38 p-chlorobenzonitrile 13 51 1,4-dicyanobenzene 34 67 This example illustrates that conversion to the thioamide impurity was suppressed in samples containing acetonitrile. Further, samples containing acetonitrile and p-methoxybenzonitrile had very little conversion to the thioamide impurity.
0.98 g (78%)
With ammonium hydroxide In ethanol
3 Preparation of 4-[2--(4--Methyl)thiazolyl]phenol
Example 3 Preparation of 4-[2--(4--Methyl)thiazolyl]phenol This compound was prepared as described by Suter, C.M., et al., J Am Chem Soc (1930) 52 :1585. 4-Methoxybenzonitrile (1.0 g 7.5 mmol) was dissolved in 6.0 ml ethanol and 2.0 ml of 29% ammonium hydroxide was added. The solution was saturated with hydrogen sulfide and the flask was closed. It was heated at 100° for 1 hr and cooled. Upon evaporation, a yellowish residue remained. This was crystallized from hot water to yield the thioanisamide as yellow leaflets, yield 0.98 g (78%) mp 148-149°.
With pyridine; hydrogen sulfide; triethylamine at 20℃; for 72h;
Multi-step reaction with 2 steps
1: trichlorothiophosphine; water; triethylamine / Neat (no solvent)
2: water / 2.5 h / 85 - 90 °C / Neat (no solvent)
Multi-step reaction with 2 steps
1: water / 90 °C / Green chemistry
2: water / 90 °C / Green chemistry
With tetraphosphorus decasulfide In ethanol at 0 - 60℃;
With sodium monohydrogen sulfide x-hydrate; magnesium(II) chloride hexahydrate In N,N-dimethyl-formamide for 6h;
93.3 g
With hydrogen sulfide; triethylamine In methanol at 80℃; for 5h; Autoclave;
3
Example 3: In a 1 L autoclave, 82.3 g (0.62 mol) of 4-methoxybenzonitrile, 185 g of methanol and 18.8 g (0.19 mol) of triethylamine were charged, a lid was put It was fixed with the attached nut to prevent pressure leakage. After raising the temperature to 80 ° C., 34.0 g (1.00 mol) of hydrogen sulfide was blown over 5 hours until a constant pressure of 0.9 MPa was reached. After the pressure meter became constant at 0.9 MPa, the mixture was stirred at 80 ° C. for 5 hours, then cooled to room temperature, and released under pressure. The interior of the autoclave was purged with nitrogen three times and the inner solution was transferred to a 2 L four-necked flask equipped with a stirrer, a condenser, a thermometer and a dropping funnel. 618 g of water and 19 g of a 35 mass% hydrochloric acid aqueous solution were sequentially added dropwise to precipitate crystals, and the crystals were filtered and washed with water.144.6 g of wet cake of 4-methoxyphenylthioamide was obtained. The obtained wet cake of 4-methoxyphenylthioamide was dried using an evaporator at 50 ° C. under a reduced pressure of 10 torr for 2 hours. 4-methoxyphenylthioamide and methanol content were measured using GC (gas chromatography), and the moisture content of the reaction solution was measured using Karl Fischer. As a result, it was 93.3 g (0.56 mol) of 4-methoxyphenylthioamide, the methanol content was 0.60 mass% based on 4-methoxyphenylthioamide, the water content was 0 to 4-methoxyphenylthioamide 0.58 mass%
Stage #1: 4-methoxybenzonitrile With sodium hydrogensulfide; 2,2'-[1,2-ethanediylbis(oxy)]bisethanol at 20℃;
Stage #2: With sulfuric acid at 110℃; for 72h;
With tetraphosphorus decasulfide In 1,4-dioxane at 20℃; for 168h;
With pyridine; diammonium sulfide; triethylamine In water at 50℃;
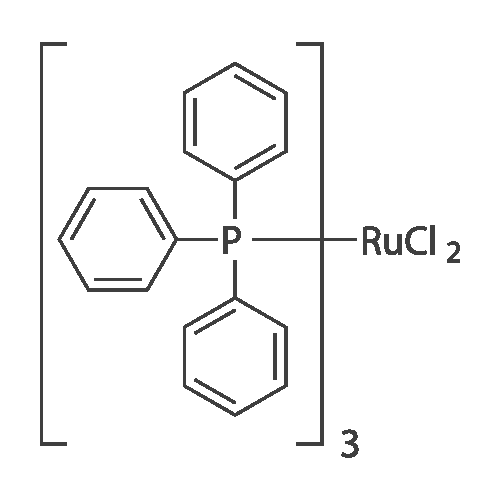
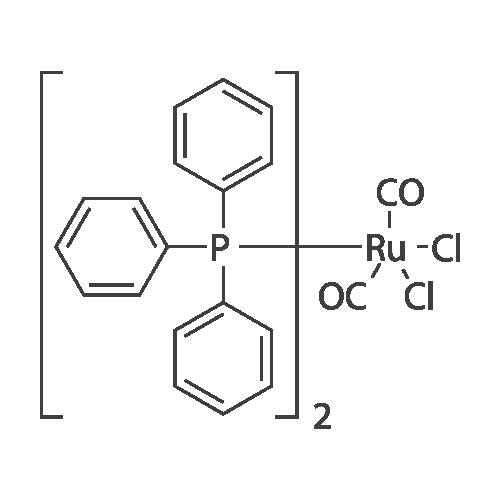
With C18H57O3P6Ru2(1+)*C6H5O(1-)*C6H6O; water; In 1,4-dioxane; for 6h;Sealed tube; Inert atmosphere; Schlenk technique;
General procedure: Complex 3 (18.3 mg, 0.0204 mmol), 1,4-dioxane (1.0 ml),benzonitrile (103.7 mg, 1.18 mmol), water (100 mul, 5.53 mmol)were added in a screw-cap tube. The reaction mixture was heatedat 100 C for 12 h. After cooling the reaction mixture to room temperature,dibenzyl and methanol (1 ml) were added to give ahomogeneous solution and the products were analyzed by GLC.
99%
With C12H24O16Ru3*2H2O; In water; at 110℃; for 13h;Schlenk technique; Inert atmosphere;Catalytic behavior;
General procedure: Hydration reactions were carried out in Schlenk tube under N2atmosphere. The reaction mixture was prepared dissolving 5 mg(6.5 lmol) of catalyst 2 in 3 mL of H2O. The mixture was degassedand 1.5 mmol of corresponding acetonitrile substrate was addedwith micropipette to stirred solution. The reaction was allowedfor heating at 110 C using oil bath or microwave-assisted heating.The isomerization reactions of allylic alcohols were conductedSchlenk tube under N2 atmosphere. The reaction mixture wasprepared dissolving 3 mg (3.9 lmol) of catalyst 2 in 2 mL ofappropriate solvent (DMF, EtOH or H2O). The mixture wasdegassed and 1 mmol of corresponding allylic alcohol substratewas added with micropipette to stirred solution. The reactionwas allowed for heating using oil bath.The reaction solutions were analyzed by regular sampling usingGC/FID (Hewlett Packard) equipped with Beta DEX 120(30 m 0.25 mm 0.25 lm) 30 m long column. The degrees ofconversion were calculated on the basis of the ratio of areas ofthe substrate material and the products determined from correspondingchromatograms. The optimization of chromatographicmethods and the calibration procedures for detection of productsas well as substrates were realized by injection of authenticcommercial samples.
94%
With [{Au(IPr)}2(mu-OH)][BF4]; water; In tetrahydrofuran; at 140℃; under 5250.53 Torr; for 2h;Sealed tube; Microwave irradiation;
In a typical reaction, [Au(IPr)(NTf2)] (13 mg, 20 mumol, 2 mol %) or [{Au(IPr)}2(mu-OH)]BF4 (17 mg, 10 mumol, 1 mol %) was added to THF (0.5 mL) in a 2 mL microwave vial in air. Benzonitrile (103 mg, 1 mmol) was added, followed by distilled H2O (500 muL). The vial was sealed and heated in the microwave for 2 h at 140 C. (7 bar). The conversion was determined by gas chromatography
General procedure: To a freshly prepared solution of LDA (1.1eq) 3-methylpyridine was added dropwise. The resulting suspension was stirred at 0C for 30min. The nitrile (1eq) was added dropwise at such a rate that the temperature did not rise above 10-15C. Stirring was continued for 60-90min at 0C. Further LDA solution (1.1eq) was added, and the reaction mixture was heated to reflux until completition of the reaction (TLC) (further heating lead to polymerization byproducts). The final reaction mixture was allowed to cool and ice-water was added. The mixture was extracted with ethylacetate and the the solvents were evaporated under reduced pressure. The brownish residue was recrystallized from methanole to obtain the pure product.
68%
LDA was freshly prepared by adding dropwise a nBuLi solution in hexane (1.5 ml, 2.5 M, 3.9 mmol) under argon to a diisopropylamine (0.54 ml, 3.9 mmol) solution in anhydrous THF (10 ml) at -5C and stirring for 20 min. Then a solution of 3-picoline (200 mg, 2.2 mmol, 1 eq) in anhydrous THF (10 ml) was added dropwise at 0C and the orange mixture was stirred for 20 min before dropwise addition of a solution of 4-methoxybenzonitrile (316 mg, 2.2 mmol, 1 eq) in anhydrous THF (10 ml). After lh at 0C, more LDA solution in THF (10 ml) was added dropwise (3.9 mmol, prepared from 1.5 ml nBuLi and 0.54 ml diisopropylamine) and the reaction was slowly warmed to r.t during lh before being heated to reflux in a water bath for 2h. After cooling, the yellow solution was quenched carefully with saturated NH4Cl (10 ml) and water (40 ml) was added. The precipitate was filtered, washed with diethyl ether and water, dried under vacuum to afford a light yellow solid (327 mg, 68%). 1H-NMR (300 MHz, CDCl3): 3.89 (s, 3H), 6.70 (s, 1H), 7.04 (d, J = 8.8 Hz, 2H), 7.14 (dd, J = 7.8, 5.1 Hz, 1H), 7.82 (d, J = 8.8 Hz, 2H), 8.00 (dd, J = 7.8, 1.3 Hz, 1H), 8.23 (dd, J= 5.1, 1.3 Hz, 1H), 12.52 (bs, 1H).
A solution of 4-methoxybenzonitrile 2 (2.1 g, 15.7 mmol), sodium methylate (86 mg, 1.57 mmol) in 20 mL of anhydrous methanol was stirred at room temperature for 48 h. Then ammonium chloride (0.84 g, 15.7 mmol) was added, and the reaction group was stirred for another 24 h the precipitate was collected by filtration, washed with diethyl ether, and then dried in vacuum to obtain 3 as a white solid (1.3 g, 44.5%), MS (ESI) m/z: 151.1 ([MH]).
With ammonium chloride; sodium methylate; In methanol;
PREPARATION 10 4-Methoxybenzamidine Monohydrochloride To a solution of 4-methoxybenzonitrile (10.0 g) in methanol (75 ml) was added sodium methoxide (0.41 g) and the reaction was stirred at room temperature for 5 days. Ammonium chloride (4.6 g) was added and the reaction was stirred overnight. The reaction was filtered and the filtrate concentrated to an oil. After the addition of ether the off-white precipitate was collected and dried to yield the title compound (3.77 g). Electrospray MS m/z 151 [M+H]+.
General procedure: A 100 mL flask was charged with 30 mL of anhydrous MeOH, 10 mmol of the arylnitrile, and 1.0 mmol of sodium methoxide. The complex was protected from moisture and stirred for 48 h. Then, 10 mmol of NH4Cl was added and stirring was continued for 24 h. Unreacted NH4Cl was filtered, and methanol was stripped from the filtrate to afford the product aryl amidine hydrochlorides, which was dissolved in 2.5 mL 8M sodium hydroxide aqueous solution and stirred for 1 h. Then chloroform (20 ml x 3) and H2O (20 ml x 3) were added successively to extract the product, and the combined organic layer was dried with anhydrous MgSO4 and then evaporated under vacuum to remove the organic solvent to give the desired arylamidine.
General procedure: Trifluoroacetic acid (4 mL) was added to a mixture of nitrile(1.0 mmol) and thiosemicarbazide (1.1 mmol). The reaction mixturewas stirred and refluxed for 6 h. Then, it was cooled to roomtemperature and aqueous ammonia was added. The precipitatedsolidwas filtered,washed with hot water and air-dried. It should benoted that the compounds f1-f35 were directly used for the nextreaction without further purification.
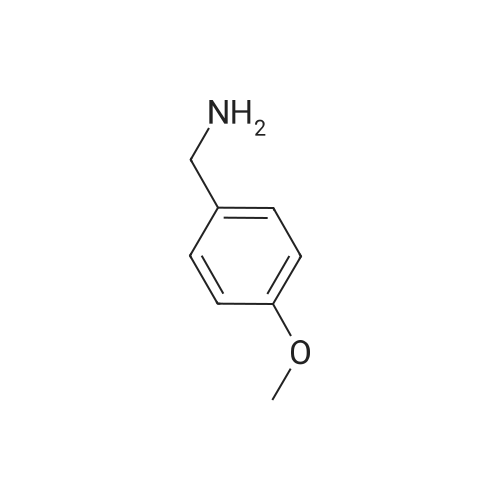
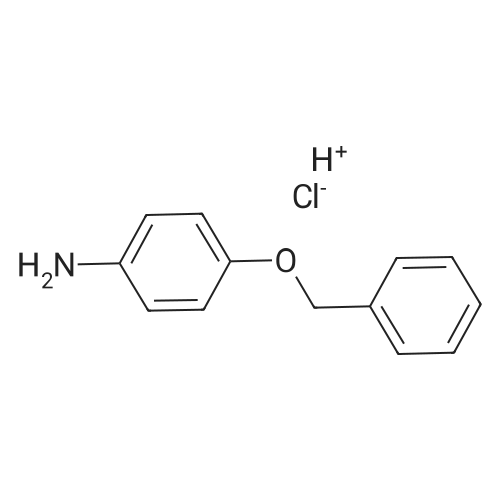
To a solution of <strong>[51388-20-6]4-benzyloxyaniline hydrochloride</strong> (3g) in tetrahydrofuran (15ML), 1. OM sodium bis (trimethylsilyl) amide in tetrahydrofuran (26. 7ML) was added dropwise at room temperature. After the mixture was stirred for 20min, anisonitrile (1.69g) was added. The reaction mixture was stirred for 4hrs, and then poured into 300ML of ice-water. The precipitate was collected by filtration, washed with diisopropyl ether to give the target compound (3.3g). 1H NMR (200MHZ, DMSO-D6, D) : 3.8 (3H, s), 5. 05 (2H, s), 6.09 (2H, bs), 6.74-6. 8 (2H, m), 6.96 (4H, d, J=8.5Hz), 7.29-7. 49 (5H, m), 7.92 (2H, d, J=8.9Hz). MS m/e : 333 (M+H) +.
With 1-methyl-3-(4-sulfonylbutyl)-1H-imidazol-3-ium trifluoromethanesulfonate; acetic acid; at 70℃; for 12h;Inert atmosphere; Ionic liquid;
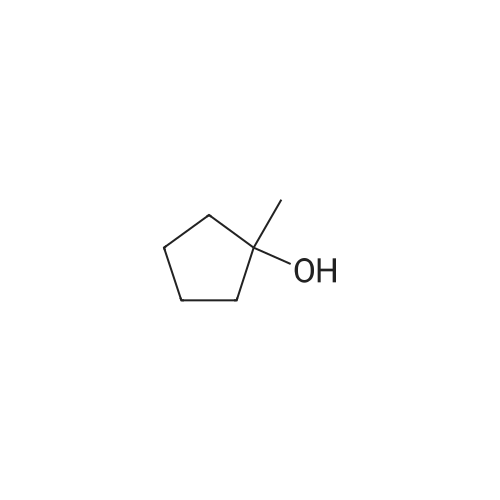
<strong>[1462-03-9]1-methylcyclopentanol</strong> (2 mmol), catalytic amounts of the ionic liquid, and 1-2 mL of acetic acid were charged into an oven-dried Schlenk tube under nitrogen. The reaction mass was stirred for 15-20 minutes at r.t. before adding the selected nitrile (1 mmol). The reaction mass was stirred at the indicated temperature for the specified time (see Table 3). The progress of the reaction was monitored by TLC and GC-MS. After completion of reaction, the reaction mass was quenched with distilled water followed by neutralization with dilute NaHCO3 solution. The product was extracted with diethyl ether, dried over anhydrous MgSO4, and the ether layer was evaporated in vacuum. The resulting crude products were chromatographed with hexane-ethyl acetate mixture (80:20) to afford pure colorless solids.
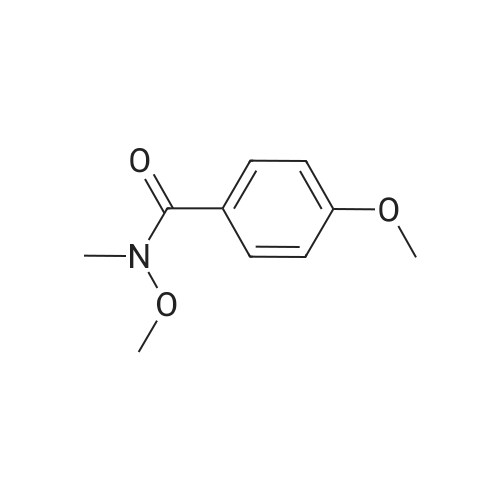
General procedure: To a solution of N,N-dimethyl benzamide (298 mg, 2 mmol) in dry THF (4 mL) was added DIBAL-H (1.04 M in hexane, 2.3 mL, 1.2 equiv) at -78 C. The mixture was stirred for 1.5 h under an argon atmosphere at from -70 C to -40 C slowly. Then, aq NH3 (concentration: 28.0-30.0%, 4 mL) and I2 (762 mg, 3.0 equiv) were added at 0 C, and the reaction mixture was stirred for 2 h at room temperature. Reaction mixture was poured into saturated aq Na2SO3 solution (10 mL) and extracted with ethyl acetate (15 mL×3). The organic layer was dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by short column chromatography on silica gel (eluent: hexane/ethyl acetate=4:1) to afford benzonitrile in 67% yield (138 mg).Most of the present prepared nitriles are commercially available and they are identified with authentic nitrile compounds.
With boron trifluoride diethyl etherate; at 20 - 80℃; for 2h;
General procedure: To a stirred solution of the corresponding epoxide 1 or oxiranes 4?7 (0.2mmol) in the corresponding nitrile (1ml) was added BF3*OEt2 (0.025ml, 0.2mmol) at room temperature. After that the mixture was stirred at 80°C for 12h. An aqueous saturated solution of sodium bicarbonate (5ml) was added and the mixture was stirred at room temperature for 5min. Then, the aqueous phase was extracted with ethyl acetate (3×5ml), and the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. Column chromatography on silica gel (60?120mesh) using chloroform/methanol (10:1) as eluent provided the desired compounds 2?3 and 8?21.
With sodium t-butanolate; In N,N-dimethyl-formamide; at 20℃; for 4h;
General procedure: A mixture of 2-substituted-1,3,4-oxadiazole (0.5 mmol), t-BuONa (0.5 mmol) and DMF or DMSO (1 mL) was stirred at room temperaturefor an appropriate time. When the reaction completed, EtOAc (5 mL) and petroleum ether (5 mL) were added, and the mixture was washed by water (3 × 10 mL) to remove DMF or DMSO. The organic layer was collected and filtered through a bed of silica gel layered over Celite.The volatiles were removed under reduced pressure to afford the final product. In a few cases, further column chromatography on silica gel was needed to afford the pure desired product.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; 2,2'-biquinoline; phenylsilane; potassium carbonate; In N,N-dimethyl-formamide; at 95℃; for 7h;Inert atmosphere; Sealed tube;
To 40 ml of DMF in the reactor, PdCl2 (dppf), organic ligand Ll, potassium carbonate, and adjuvantAnd 11 mmol of the compound of formula (I) and 13 mmol of the compound of formula (II) in the above reaction scheme, introducing nitrogen to form an inert protective atmosphere, Sealed reaction at 95 C for 7 hours, the reaction is completed by natural cooling, vacuum concentration, the use of petroleum ether and ethyl acetate mixtureSolvent (volume ratio of 1: 2) to obtain the compound of the formula (III) in a yield of 98.1%;Wherein the molar amount of the PdCl2 (dppf) is 0.6% of the molar amount of the compound of the formula (I), and the ligand L1Is used in an amount of 2.5% by mol based on the molar amount of the compound of formula (I), the molar amount of said potassium carbonate being a molar amount of the compound of formula (I)30% and the molar amount of said auxiliary PhSiH3 is 2% by mol of the compound of formula (I).
72%
With C39H32Cl2N5PRu; potassium tert-butylate; In tert-Amyl alcohol; at 130℃; for 2h;Sealed tube;
General procedure: To an oven-dried 15 mL sealed tube were added 2-aminophenylmethanol 1 (0.425 mmol), benzonitrile 2(0.25 mmol), Ru cat. b (1.94 mg, 1 mol%), and KOtBu (14.02 mg, 0.5equiv) intamyl alcohol (1 mL) under an air atmosphere. The sealedtube was capped and heated at 130C for 2 h. The reaction mixturewas cooled down to room temperature and directly concentratedunder vacuum. The crude mixture was puried by preparative thin-layer-chromatography (petroleum ether/ethyl acetate 20/1) togive the desired product 3 or 4.
Stage #1: 1-bromo-4-methoxy-benzene With TurboGrignard In tetrahydrofuran; 1,4-dioxane at 0℃; for 48h;
Stage #2: 2,2-dimethylmalononitrile In tetrahydrofuran; 1,4-dioxane at 0 - 23℃; for 0.5h;
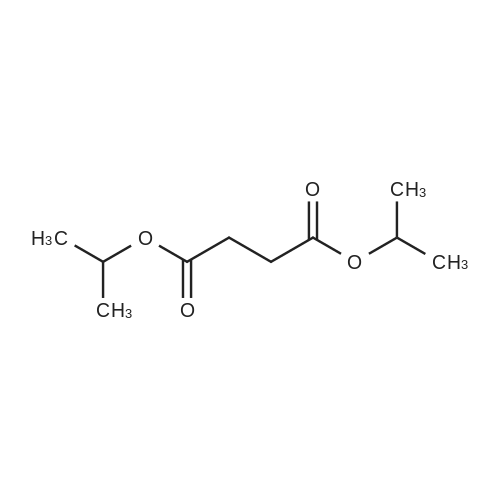
General procedure: Under nitrogen condition, potassium tert-butoxide (2.25 g, 0.02 mol) was dissolved in150 mL of tert-amyl alcohol for 15 min and the mixture was heated to 110. Iron(III) chloride (s.a) was added and stirred for 30 min then 4-chlorobenzonitrile (1.5 g, 0.01 mol) was added and stirred for 1 hr at the same temperature. Diisopropyl succinate (1.01g, 0.005 mol) was dissolved in 30 mL of tert-amyl alcohol and dropwised to the mixture for 3 hr. Then 60 mL of water was poured into mixture and stirred for 20 hr at the same temperature. After reaction, the mixture was cooled to room temperature. The mixture was filtered off and washed by water, methanol. The red pigment was purified by 50 mL methanol and stirred for 30 min at 60. After stirring, the solution was filtered off and dried under vacuum to obtain Pigment.
1-(4-methoxyphenyl)-6-methyl-6H-isoxazolo[4,3-e]-indazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
17%; 45%
With potassium hydroxide; In methanol; for 48h;Reflux;
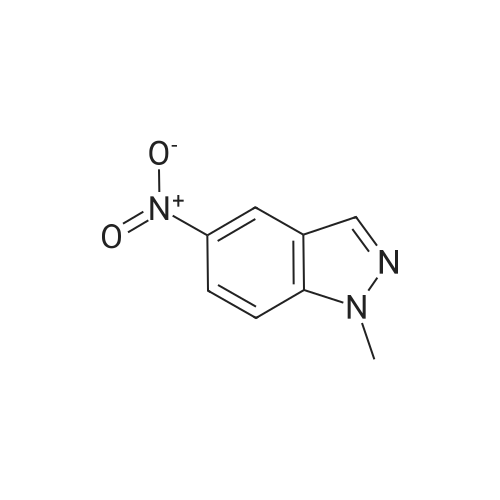
General procedure: 1-Alkyl-5-nitro-1H-indazole 1 (1.77 g, 10 mmol) andnitrile 2a-e (12 mmol) were added with stirring to asolution of KOH (30 g, 535 mmol) in methanol (70 ml).The mixture was refluxed for 48 h and then poured intowater. The precipitate was filtered off, washed with water,and air-dried to give crude product 5a-e with admixture ofside product 4a-e. Washing the crude product withacetone, evaporation of the filtrate, and recrystallization ofthe residue from MeOH gave pure compound 5a-e, whilecrude compound 4a-e remained as precipitate on the filter.Compound 4a-e was purified by recrystallization fromEtOH
3-(4-methoxyphenyl)-1,5-diphenyl-1H-1,2,4-triazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
With copper(l) iodide; 1,10-Phenanthroline; zinc(II) iodide; In chlorobenzene; at 130.0℃; for 36.0h;Sealed tube;
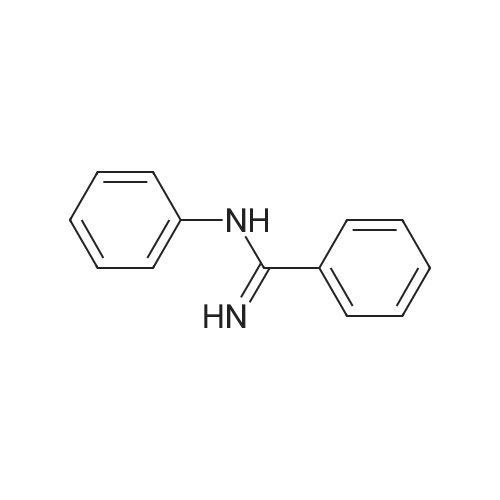
General procedure: A sealed tube equipped with a stirrer bar was charged with <strong>[1527-91-9]N-phenylbenzamidine</strong>(1a; 0.0393 g, 0.2 mmol), benzonitrile (2a; 41 muL, 0.40mmol), CuI (0.0190 g, 0.10 mmol), ZnI2 (0.0063 g, 0.02 mmol),phenanthroline (0.0040 g, 0.02 mmol) and chlorobenzene (1 mL). The resulting mixture was stirred at 130 C for 36 h. After reaction completion,the residue was directly purified by flash column chromatography(EtOAc-petroleum ether) to afford pure product 3aa.
With potassium tert-butylate; copper(ll) bromide; In tert-butyl alcohol; at 20℃; for 24h;
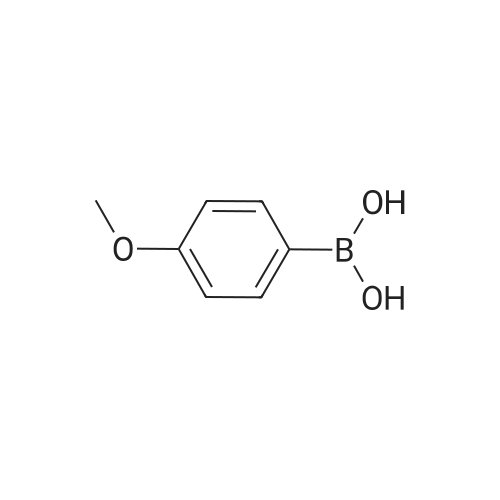
General procedure: A 10 mL round-bottomed flask was charged with the appropriate nitrile 2 (0.50 mmol), arylboronic acid 1(0.60 mmol), CuBr2 (6 mg, 5mol%), t-BuOK (168 mg, 1.50 mmol), and t-BuOH (3.0 mL), and the mixture was stirred at r.t. until the reaction was complete (TLC). H2O (4.0 mL) was added, and the mixture was extracted with EtOAc (3 ×10 mL). The combined organic layers were washed twice with H2O,dried (Na2SO4), and concentrated to give a residue that was purified by column chromatography (silica gel, PE-EtOAc).
N-(benzo[d][1,3,2]dioxaborol-2-yl)-N-(4-methoxybenzyl)benzo[d][1,3,2]dioxaborol-2-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With C29H28Cl2N2Ti; In neat (no solvent); at 65℃; for 10h;Schlenk technique; Inert atmosphere; Glovebox;
General procedure: Catalyst 1 (10 mol%) and catecholborane (0.44mmol) wereadded to a Schlenk flask inside the glove box, followed by organicnitriles (0.2 mmol). The reaction mixture was heated continuouslyat 65 C under neat condition or under toluene for a stipulated time,as mentioned in Table 2. The progress of the reaction was monitoredby 1H NMR spectroscopy using 1,3,5-trimethoxybenzene(50 mol%) as an internal standard.
With potassium phosphate; copper(l) iodide; In water; at 80℃; for 12h;Schlenk technique;
96 mg (0.5 mmol) of phenyl diazonium tetrafluoroborate, 0.1 mmol of cuprous iodide, 0.5 mmol of potassium phosphate and 1.0 mmol of water are Under a nitrogen atmosphere, 1 mL of 4-methoxybenzonitrile was added to the Schlenk tube under air conditions at 80 C. (stirring reaction for 12 hours resulted in a yield of 73%.
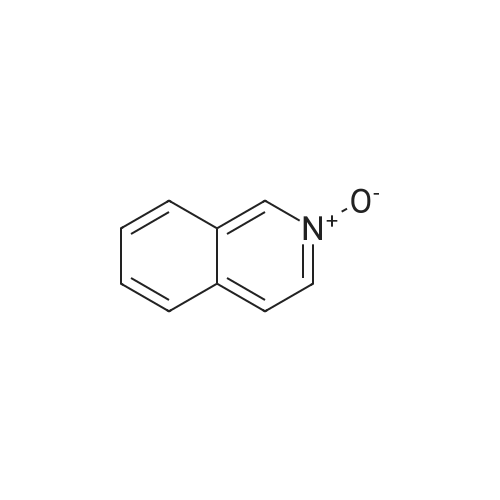
With toluene-4-sulfonic acid; In water; toluene; at 110℃; for 12h;Inert atmosphere;
The raw material quinoline-N-oxide (0.2 mmol, 1 equiv) and p-toluenesulfonic acid (TsOH, 0.6 mmol, 3 equiv) were added to the reaction vessel, and the reaction vessel was continuously subjected to three pumping-nitrogen charging operations, and then continued. To the reaction vessel were added toluene (Toluene) and water (H2O) (3:1 = 2 mL) and the starting material 4-methoxybenzonitrile (0.45 mmol, 1.5 equiv), followed by a reaction temperature of 110 C in an oil bath. Stir under the reaction until the end of the reaction (about 12h), Then, the reaction mixture was diluted with ethyl acetate (10 mL), and the diluted solution was transferred to a separating funnel, extracted with saturated brine, and the aqueous and organic phases were separated, and the aqueous phase was extracted with ethyl acetate. Then, all the organic phases were combined, and then the combined organic phase was dried by adding anhydrous sodium sulfate (5 g). After 5 min, the filter cake was washed with ethyl acetate (5 mL×3×), then concentrated under reduced pressure, and finally, the concentrate was separated by column chromatography (with petroleum ether and ethyl acetate in a volume ratio of 10:1 as eluent). The eluate was collected, the solvent was spun off, and after separation and purification, a white liquid was obtained with a yield of 87%.
With [2,2]bipyridinyl; bis(acetylacetonate)nickel(II); 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclo-hexadiene; at 80℃; for 24h;Inert atmosphere;
2.6 mg (10 mmol) of nickel (II) acetylacetonate (manufactured by Sigma-Aldrich), 2,2'-bipyridine (manufactured by Tokyo Chemical Industry Co., Ltd.) 1 , 18.0 μL (100 mmol) of <strong>[66107-29-7]4-methoxyphenyl triflate</strong> (manufactured by Tokyo Chemical Industry Co., Ltd.) were added and dissolved in 3.0 mL of acetonitrile (manufactured by Kanto Kagaku Co., Ltd.). 706 mg (250 mmol) of Me4BTDP was added, and the mixture was stirred under an Ar atmosphere at 80 C. for 24 hours. Thereafter, 3.7 mg (20 mmol) of ferrocene (manufactured by Tokyo Chemical Industry Co., Ltd.) was added as an internal standard, the reaction solution was concentrated and 1 H NMR (400 MHz, CDCl 3) measurement was carried out,The yield of 4-methoxybenzonitrile was 64%.
With carbonyl bis(hydrido)tris(triphenylphosphine)ruthenium(II); palladium diacetate; sodium hydride; silver(l) oxide; 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazole trafluoroborate; In toluene; at 110℃; for 18h;Inert atmosphere; Schlenk technique;
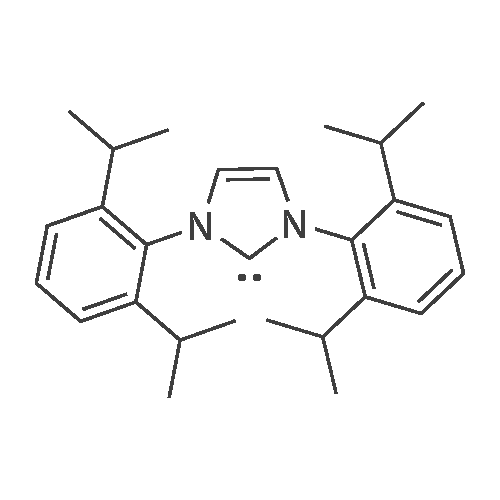
Under the protection of nitrogen,Add to a 10ml Schlek reaction tube (a glass instrument commonly used in anhydrous anaerobic operation)1.0 mmol p-methylbenzyl alcohol,1.2mmol 4-methoxybenzonitrile,0.08mmol RuH2(CO)(PPh3)3,0.1 mmol palladium acetate,0.12 mmol of 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazole tetrafluoroborate,0.1 mmol of silver oxide, 3.0 mmol of sodium hydride,And 5ml of toluene,Replace the reaction tube three times with nitrogen,Then heated to 110 C with an oil bath under magnetic stirring.The reaction was refluxed for 18 hours.Remove the oil bath,Drop to room temperature; add 3 ml of water to the reaction solution,Extract three times with 5 ml of dichloromethane.The organic phases were combined and dried over anhydrous MgSO4 for 30 min.Filtration; the filtrate was concentrated using a rotary evaporator.The concentrated solid is treated with dichloromethane as a solvent.Recrystallization gave pure product 30, yield 89%.
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; ammonium acetate; oxygen; nitric acid; In acetic acid; at 50℃; for 20h;Autoclave;
General procedure: The reactions were carried out a ~40 mL, Teflon-lined, stainlesssteelautoclave. AcOH (2 mL), TEMPO (0.25 mmol), HNO3 (0.25mmol), the appropriate benzylic methyl ether (0.5 mmol), andNH4OAc (1.5 mmol) were added sequentially to the autoclave.Subsequently, the autoclave was pressurized to 1 MPa with O2,and the reaction mixture was heated with magnetic stirring at50 C for 20 h, then cooled to r.t. The mixture was then dilutedwith Et2O (15 mL) and H2O (5 mL), and adjusted to pH 7-8 with2 M aq NaOH. The two layers were separated, and the aqueouslayer was extracted with Et2O (3 × 15 mL). The organic layerswere combined, dried (Na2SO4), filtered, and concentrated to avolume of approximately 3 mL in a rotary evaporator. GC analysisof the concentrated organic phase, with biphenyl or 1,2,4,5-tetramethylbenzene as internal standard, provided the GC yieldof the product. The crude product in the concentrated organicphase from another parallel experiment was purified bycolumn chromatography [silica gel (200-300 mesh), EtOAc-PE].
Stage #1: methyllithium With cerium(III) chloride In tetrahydrofuran at -78℃; for 0.5h; Inert atmosphere;
Stage #2: 4-methoxybenzonitrile In tetrahydrofuran at -78 - 20℃; Inert atmosphere;
Stage #3: With ammonium hydroxide In tetrahydrofuran at 20℃; for 1h; Inert atmosphere;
Synthesis of 21
Step A: Anhydrous CeCl3 (0.84 g, 3.4 mmol) was stirred in dry THF (10 mL), at room temperature, under a N2 atmosphere for 2 h. Then, the solution was cooled at -78 °C, followed by dropwise addition of MeLi (2.2 mL, 3.4 mmol, 1.6 M), and allowed to stir for 30 min at -78 °C. Then, a solution of 4-methoxybenzonitrile (0.15 g, 1.1 mmol) in dry THF (5 mL) was added dropwise to the above solution. The crude reaction was stirred at room temperature overnight, quenched by adding 1.5 mL of concentrated NH4OH, and allowed to stir for 1 h at room temperature. The organic layer was filtered, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was used for next reaction without further purification.
With zinc perchlorate; In neat (no solvent); at 50℃; for 1h;
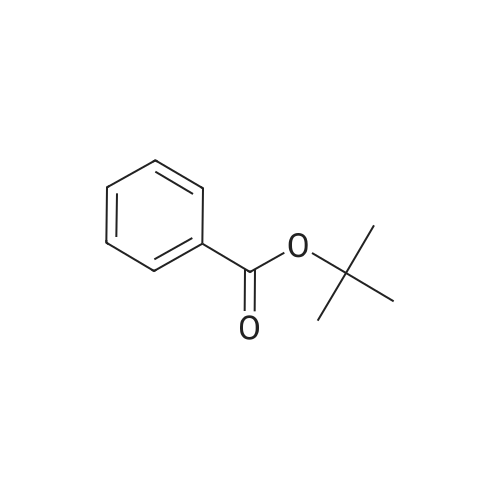
General procedure: A mixture of nitrile (5 mmol), <strong>[774-65-2]ter<strong>[774-65-2]t-butyl benzoate</strong></strong> (5.5 mmol) and Zn(ClO4)2·6H2O (2 mol%) was placed in around bottom flask. Then, the reaction mixture was heated at 50 C for the given time. After completion of the reaction monitored by thin layer chromatography (TLC), the reaction mixture was quenched with 5-ml water. Then the reaction system was added 10 ml aqueous NaOH solution (1 mol/L) and continued to be stirred 5 min and extracted with ethyl acetate (3 × 10 ml). The organic layers were collected, combined, washed with water (3 × 10 ml), driedover anhydrous Na2SO4, and concentrated under vacuum.
With C39H32Cl2N5PRu; potassium tert-butylate; In tert-Amyl alcohol; at 130℃; for 2h;Sealed tube;
General procedure: To an oven-dried 15 mL sealed tube were added 2-aminophenylmethanol 1 (0.425 mmol), benzonitrile 2(0.25 mmol), Ru cat. b (1.94 mg, 1 mol%), and KOtBu (14.02 mg, 0.5equiv) intamyl alcohol (1 mL) under an air atmosphere. The sealedtube was capped and heated at 130C for 2 h. The reaction mixturewas cooled down to room temperature and directly concentratedunder vacuum. The crude mixture was puried by preparative thin-layer-chromatography (petroleum ether/ethyl acetate 20/1) togive the desired product 3 or 4.
With cesium fluoride; lithium hexamethyldisilazane; In tetrahydrofuran; at 110℃; for 12h;Green chemistry;
General procedure: Lithium bis(trimethylsilyl)amide (66.8 mg, 0.4 mmol) and cesium fluoride (30.4 mg, 0.2 mmol) were placed in a microwave tube in a glove box. Add 0.4 mL of cyclopentyl methyl ether, Then <strong>[95-52-3]2-fluorotoluene</strong> (66 muL, 0.60 mmol) and benzonitrile (20 muL, 0.20 mmol) were added separately using a micro syringe. which was taken out from the glove box and refluxed at 110 C for 12 hours. After cooling to room temperature, the reaction was capped and three drops of water were added to quench the reaction. The solvent was removed under reduced pressure and the crude product was purified by column chromatography ( petroleum ether: ethyl acetate = 20:1) to give 2-phenylindole (34.7 mg) , 90% yield).
With tert.-butylnitrite; iodine; In neat (no solvent); at 25℃; for 5h;
General procedure: To a round-bottom flask (25 mL) was added phenylhydrazine hydrochloride 1a (43 mg, 0.3 mmol), benzonitrile 2a (0.3 mL, 3 mmol), t-BuONO (53 μL, 0.45 mmol), I2 (38 mg, 0.15 mmol), the mixture was well stirred for 6 h at 25 oC (the whole process was closely monitored by TLC). Then the reaction mixture was purified by a flash silica gel column chromatography (eluent: Petroleum ether (PE)/Ethyl acetate (EA) = 10:1) to give N-phenylbenzamide 3a as a white solid (38 mg, 64%).
With hydroxylamine hydrochloride; In methanol; at 120.0℃; for 12h;Inert atmosphere; Sealed tube;
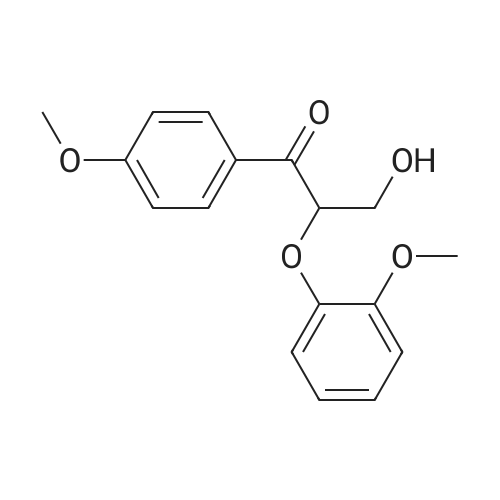
In a 15 mL pressure bottle, add 1 mL of methanol, 0.1 mmol of <strong>[92409-23-9]3-hydroxy-2-(2-methoxyphenoxy)-1-(4-methoxyphenyl)propyl-1-one</strong>, and 0.3 mmol of hydrochloric acid. Hydroxylamine was filled with nitrogen to replace the atmosphere, sealed, and stirred at 120 C for 12 h. After the reaction was completed, the product was separated by centrifugation and the chromatogram was determined. The conversion and the yield are shown in Table 1.
21%; 56%; 19%
With hydroxylamine hydrochloride; triethylamine; In methanol; at 120.0℃; for 12h;Inert atmosphere; Sealed tube;
In a 15 mL pressure bottle, add 1 mL of methanol, 0.1 mmol of <strong>[92409-23-9]3-hydroxy-2-(2-methoxyphenoxy)-1-(4-methoxyphenyl)propyl-1-one</strong>, 0.3 mmol of hydrochloric acid. Hydroxylamine and 0.2 mmol of MgO were filled in a nitrogen-substituted atmosphere, sealed, and stirred at 120 C for 12 h. After the reaction was completed, the product was separated by centrifugation, and the conversion and the yield were shown in Table 1.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






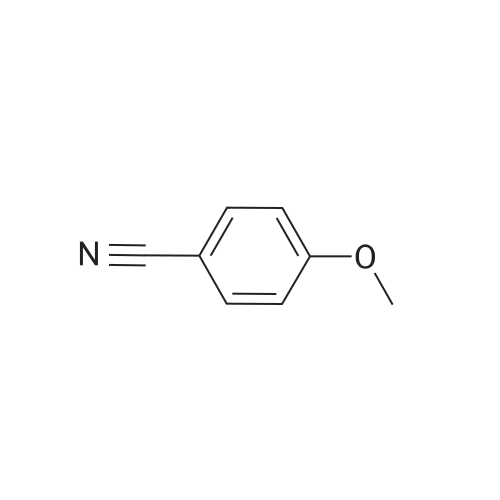

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping