
Abstract
AlN particles are formed in liquid Al metals/alloys during liquid-dealing and casting. They may act as potential nucleation sites during solidification. Along its [0 0 0 1] axis, AlN consists of Al3+N3- dipolar double-atom layers in the ionic model. Thus, the AlN{0 0 0 1} substrates are terminated by either an Al3+ or a N3- layer, being polar. Here we investigate the atomic ordering at the interfaces between liquid-aluminum and AlN{0 0 0 1} using an ab initio molecular dynamics technique. We have observed a rich variety of interfacial chemistry and identified an ordered Al layer terminating the substrates. The newly formed terminating Al atoms are positively charged. The liquid Al adjacent to the interfaces exhibit strong layering but weak in-plane ordering. The obtained information helps get insight into the role of aluminum nitride as potential nucleation sites in solidification of Al-metals, and further enriches our knowledge about nucleation
Similar content being viewed by others

1 Introduction
Both aluminum oxide and nitride particles are formed during liquid-dealing and casting of Al metals/alloys.[1,2,3,4,5] Yan and colleagues investigated secondary phases and their interfaces with the Al matrix in the Al-2Mg-2Si-0.25Cu alloy sintered in nitrogen and observed hexagonal AlN nano-crystallites in the prepared samples.[1] Fale, et al. found formation of AlN particles in Al metals by means of nitridation via decomposition of ammonia chloride in liquid Al. The formed AlN particles in the Al matrix exhibit more plate-like, indicating dominant AlN{0 0 0 1} surfaces.[2] AlN particles in cast Al metals of commercial purity were observed to exhibit a short rod-like morphology with an average length of 375nm and average width of 104nm and the most close plane {0 0 0 1} being the exposed plane.[3] Recent experiments also revealed co-formation of aluminum nitride and oxide particles in Al-3Fe alloys.[4] The native AlN particles have mostly short-rod- or plate-like morphologies with the dominant {0 0 0 1} facets. They have nontrivial impacts on the mechanical performance of the cast parts, and may also compete with the oxide particles as potential nucleation sites during solidification.[3,4,5]
The early-stage solidification theory[5,6] suggests that at temperature above the nucleation temperature, there is atomic ordering in the liquid metal adjacent to the solid-substrate. This is referred as prenucleation,[5,6,7,8] which produces a precursor for following nucleation and grain initiation.[6,9] Thus, knowledge about atomic ordering at the interfaces between liquid Al and the AlN{0 0 0 1} substrates (L-Al/AlN{0 0 0 1} in short) is important to understand the role of the native AlN and oxide particles as potential nucleation sites in solidification of Al-metals/alloys.[2,3,4,10,11]
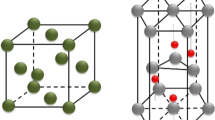
AlN has the Wurtzite-type structure with a hexagonal lattice (Figures 1(a) and (c)).[12] Along its [0 0 0 1] axis the structure is composed of AlN double-atom layers. Each Al/N is tetrahedrally coordinated by N/Al: three in the double-atom layer and one to the neighboring layer. Previous electronic structure study showed that AlN exhibits ionic nature,[13] as illustrated in the electron density distribution (Figure 1(b)). The frame of the structure is electronically determined by the N-sublattice.[13] It is, thus reasonable to separate them between the AlN doble-atom layers to create the AlN{0 0 0 1} substrate-surfaces. The obtained AlN{0 0 0 1} substrates are terminated by either an Al3+ or a N3- layer, being polar. Such polar surfaces are unstable in ambient conditions.[14,15] However, it is different in liquid metals as the free electrons of metals compensate the substrates’ charging.[10,11] Knowledge about the L-Al/AlN{0 0 0 1} interfaces can enrich our understanding about nucleation.[6,8,16]

(Color-online)The schematic structure of AlN with the wurtzite-type (a), the related electron density distribution from our DFT-GGA calculations (b) and the projection along its [0 0 0 1] axis (c). The red lines in (a) and (c) and the green in (b) represent the axis of the unit cell. The dark spheres represent N and silvery Al. The yellow color in (b) represents the iso-surfaces with ρ(r0) = 0.035 e/ Å3
Experimental efforts have been made to understand the formation and morphologies of the AlN particles in liquid Al metals/alloys,[1,2,3,4] wetting of liquid Al on AlN substrates[17] and role of AlN in Al matrix for nano-composite materials.[18,19,20,21,22] High-resolution transmission electron microscopy images revealed the orientational relations between AlN and solid Al, AlN{0 0 0 1}/α-Al{1 1 1} for cast samples.[4,23] Theoretical efforts have been made on its bulk and the surfaces of AlN[13,16,24,25] and interfaces between AlN and solid metals including aluminum.[25,26,27,28,29] Recently, theoretical efforts have also been made on understanding of liquid-metal/solid-substrate interfaces.[30,31,32,33,34,35,36] Semi-empirical atomistic molecular dynamics simulation approaches were employed for investigation of liquid-metal/solid-metal interfaces,[30,31,32] impacts of lattice misfit on atomic ordering of liquid near the solid substrates[7,31] and effects of the surface roughness of substrates on atomic ordering at the liquid-metal/solid-metal interfaces.[33] Parameter-free ab initio molecular dynamics simulations (AIMD) were successfully performed for the L-Al/TiB2 and L-Al/TiAl3 interfaces.[34,35,36] Recent attention has been paid to interfaces between liquid metal and oxide substrates.[10,11,37,38,39,40,41,42] Here, we investigate prenucleation at the L-Al/AlN{0 0 0 1} interfaces using an AIMD method. We reveal formation of ordered Al layers terminating the substrates. The Al adjacent to the substrates exhibit unusually strong layering but weak in-plane ordering. The obtained information here is helpful to get insight into not only the role of AlN particles in heterogeneous nucleation of Al-based metals/alloys, further into designing of new grain refiners, but also the chemistry of AlN-containing nano-composites.[18,19,20,21,22,27,28,29]
2 Methods
In our AIMD simulations periodic boundary conditions are utilized. We built a hexagonal supercell with a = 5 a0, where a0 is the length of a-axis of the hexagonal AlN unit-cell with consideration of the thermal expansion at the simulation temperature.[12] The length of the c-axis is determined by the thickness of the AlN slab and the volume of the Al atoms with the density at the simulation temperature.[43] Thus, a supercell with a = 15.63 Å and c = 48.60 Å was built for the L-Al/AlN{0 0 0 1} interfaces. This cell contains in total 700 atoms: 550 Al and 150 N atoms. Three different interfaces were built, Case_A: The AlN substrate is terminated by an Al- and a N-layer, respectively; Case_B: Both substrate surfaces are N-terminated with an Al-Al anti-phase structure in the substrate; and Case_C: Both substrate surfaces are terminated by an Al-layer with a N-N antiphase in the substrate. The supercell is deliberately large, that helps obtain statistically meaningful results and avoid risk of artificial crystallization of the liquid.
We employ a plane-wave pseudo-potential approach (code VASP, Vienna Ab initio Simulation Package).[44] VASP utilizes the first-principles density functional theory (DFT) within the projector-augmented wave (PAW) framework.[45] The exchange and correlation terms are described using the Generalized Gradient Approximation (GGA-PBE).[46] We use cut-off energies, ECUT/EAUG = 450.0 eV/650.0 eV which are higher than the default values (EMAX/EAUG = 240.0 eV/291.1 eV for Al and 400.0 eV/627.1 eV for N, respectively). For the AIMD simulations, we employed a cut-off energy of 320 eV which is higher than the EMIN values of the pseudopotentials of the related atoms,[44] and the Γ-point.[47] The latter is due to the lack of periodicity of the whole systems.[8,37,38,39,40,41,42] Test simulations from 200 eV to 400 eV showed the present cut-off energy is reasonably good.
The liquid Al samples were generated by equilibrating for about 10 ps at 3000 K. Then, they were cooled to the designed temperature (1000 K) to eliminate excess internal stress and defects created at the high temperature. We used the produced liquid Al samples together with the AlN{0 0 0 1} slabs for building the L-Al/AlN{0 0 0 1} interfaces. We also used a two-step approach: We first performed AIMD simulations with the substrate atoms pinned for 2.8 ps (1.5 fs per step). Then, we equilibrated further the systems with full relaxation of the atoms for another 4000 to 7000 steps. Analysis of the configurations showed no further increase of atomic ordering at the interfaces. The two-step approach avoids risk of possible collective atomic movements occurring when we relax all atoms from start. The time-averaged method was used to sample the interfaces over about 3.0 ps to ensure statistically meaningful results.[41,48]
3 Results
Structural optimizations using above settings produced lattice parameters, a = 4.039 Å for the cubic α-Al (experimental value 4.0325 Å at 0 K[43]) and a = 3.125 Å, c = 5.008 Å for the hexagonal AlN (experimental values, a = 3.1115 Å, c = 4.9798 Å at room temperature[12]). It is not unusual for the density-functional theory within the GGA approximation to overestimate the lattice parameters of crystals.[46,49] Overall, the first-principles calculations reproduced the experimental values well (within 1 pct).
3.1 Atomic Evolutions at the L-Al/AlN{0 0 0 01} Interfaces
During the AIMD simulations, liquid Al atoms move to the pinned AlN substrates and gradually form stable structures. Correspondingly, the total-valence electrons energies of the interfaces decrease quickly at first and then slowly. It reaches equilibrium after about 1 ps as shown in Figure 2 for Case_A as an example. Full relaxation of the Al and N atoms in the substrates enhances their kinetic energy of the substrate atoms. Correspondingly, the local structures are distorted and the energy of the system increases. The system reached again thermal equilibrium within about 0.5 ps after full relaxation (Figure 2) and the liquid Al atoms near the substrates become ordered (Figure 3). Simulations for the other cases showed some differences. For the interface with Al-terminations (Case_C), fully atomic relaxation induced re-construction of the AlN substrate and the N-N antiphase disappeared. Consequently, the system became Case_A. To realize a system with the two Al-termination surfaces we fixed the anti-phase N atoms in the inner substrate. The simulations revealed instability of the system that on one substrate, the N atoms in the substrate diffuse into the liquid. Fortunately, on the other substrate, the AlN substrate is still stable and the nearby atomic arrangements of liquid are similar to those at the corresponding substrate in Case_A. The simulations showed that the atomic arrangements of the N-terminated substrate (Case_B) is similar to that at the N-terminated side of Case_A. Thus, we discuss the Case_A in the rest of this paper.

Variation of the total-valence-electrons energy of the L-Al/AlN{0 0 0 1} (Case_A) on time during the AIMD simulations at 1000 K. The vertical dotted line represents the start of full relaxation

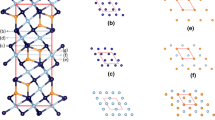
(Color-online)Snapshot of the equilibrated L-Al/AlN{0 0 0 1} interfaces at 1000 K (upper) and the atomic density profile (below). The dark spheres represent N and the silvery Al. The peak at about 13.1 Å indicated by a red line with arrow belongs to the terminating Al layer to Surface 1(S1) and the peak at about 46.5 Å belongs to the terminating Al layer of substrate surface 2 (S2)
3.2 Atomic Ordering at the L-Al/AlN{0 0 0 01} Interfaces
Figure 3 shows the snapshot of the equilibrated interfaces (Case_A). The atoms in the AlN substrate are ordered, being structurally similar to that of the bulk (Figure 1a), whereas the Al away from the substrate display disordering, being liquid-like. The liquid Al atoms near the substrates show density variation along its z-axis. This phenomenon is referred as layering which can be assessed by the atomic density profile, ρ(z),[7,8,30,31]
here, Lx and Ly are the in-plane x and y dimensions of the cell, respectively, and z the dimension perpendicular to the substrate. Δz is the bin width (here is 0.2 Å ), and Nz(t) is the number of atoms between z - (Δz/2) and z + (Δz/2) at time t. ‹Nz(t)› means a time-averaged number of atoms in the duration. The ρ(z) for L-Al/AlN{0 0 0 1} using the summed configurations over 3 ps was obtained via Eq. [1] and is plotted in Figure 3.
The atomic density profile of the AlN substrates shows sharp peaks, each of which contains a double-subpeak, corresponding well to the atomic arrangements of the substrate. There is an Al layer terminating the substrates and five more recognizable Al layers at both interfaces. The peak heights of the Al layer decreases with the distance from the substrate surfaces (Figure 3). There are subtle differences at the two L-Al/AlN interfaces.
At the substrate surface 1 (S1 in Figure 3) the termination Al layer is close to the outmost N layer. The 1st Al layer has its peak 2.40 Å from the terminating Al layer. At S2, the terminating Al peak is 2.0 Å away from that of the N-peak of the substrate (Figure 3). At both interfaces, the inter-layer spacing between the Al layers in the liquid varies slightly (~ 0.1 Å ). The 1st Al layer is well-separated from the terminating Al layer, but admixed with the 2nd Al layer. The rest Al layers admixed with each other. The peak height of the 5th Al layer is 0.062 / Å3) closer to the density of liquid Al, being 0.057 (1/ Å3).
The epitaxial nucleation model suggests a layer-by-layer growth, and that the substrate surface templates nearby liquid to nucleate.[9] Knowledge about the atomic arrangements at the layers adjacent to the substrate determine is crucial. Figure 4 shows the atomic arrangements integrated over 3 ps at the Al layers adjacent to the substrates.

(Color-online)Summed configurations of the Al layers over 3 ps at the L-Al/AlN{0 0 0 1} interfaces. The silvery spheres represent Al and the red lines represent the in-plane axis. On top, (a) represents the atomic arrangements of the terminating Al layer, (b) the 1st Al layer and (c) the 2nd Al layer. The row (I) is for the surface 1 (S1) and (II) surface 2 (S2)
To assess the atomic ordering at an individual layer, we employed atomic in-plane ordering coefficient, S(z)[7,8,30,31]:
here, the summation is over all atoms within a given bin of width. Q is the reciprocal lattice vector, rj is the Cartesian coordinates of the jth atom, and Nz is the number of atoms in the layer. S(z) assesses the atomic ordering in an individual layer. We analyzed the configurations over 3 ps according to Eq. [2]. The results are plotted in Figure 5. The in-plane ordering coefficient at S1 is notably higher than that at S2. It decays quickly at both interfaces. The 1st Al layers at both interfaces exhibit little atomic ordering.

In-plane ordering coefficients of atomic layers at the L-Al/AlN{0 0 0 1} interfaces. The meaning of the surfaces S1 and S2 are shown in Fig. 3. The 0 at x-axis represents the terminating Al layer, 1 the 1st, 2 the 2nd and 3 the third Al layer. The lines linking the points are used to guide readers’ eyes
Statistical analysis showed that the number of atoms at the termination Al layers at both L-Al/AlN{0 0 0 1} interfaces is almost equal to that of the N layers at the substrate. Note that the lattice misfit between Al{1 1 1} and AlN{0 0 0 1} is −7 pct. This means that strong interfacial interaction overcomes the lattice misfit. Meanwhile, analysis revealed the higher ratios of Al atoms at the layers in the liquid to that of the termination layer: (N(nth-layer)/N(Alterm) = 1.10 for the 1st, 1.13 for the 2nd and 1.15 for the 3rd Al layer at the S1 interface; (N(nth-layer)/N(Alterm) = 1.02 for the 1st, 1.13 for the 2nd and 1.16 for the 3rd Al layer at S2, respectively. The higher numbers of Al atoms at the liquid layers correspond to the (negative) lattice misfit.
To get a better understanding on the origin of the atomic arrangements of the terminating Al layers at the two AlN substrate surfaces, we analyzed the local chemical bonding and polyhedral coordination of N at the L-Al/AlN{0 0 0 1} interfaces in Figure 6.

(Color-online)Tetrahedral coordination of the outmost N atoms by Al at the L-Al/AlN{0 0 0 1}S1 (a) and L-Al/AlN{0 0 0 1}S2 (b) interfaces. The dark brown spheres represent N and silvery Al
Figure 6 shows tetrahedral coordination of the outmost N atoms by Al at both interfaces. This corresponds to the strong Al-N bonding and high stability of AlN (melting point of AlN is about 2200 °C). The terminating Al atoms can thus, be considered as extension of the substrates. At L-Al/AlN{0 0 0 1}S1, the terminating Al atoms have three N neighbors and correspondingly display high ordering (Figure 4), whereas the terminating Al atoms at S2 have just one N neighbor, being more flexible and display larger spacing to the outmost N and higher mobility.
In brief, the terminating Al atoms are strongly bonded to the AlN substrates. The layering phenomenon at both L-Al/AlN{0 0 0 1} interfaces is pronounced. The Al atoms in the liquid adjacent to the substrates form flat peaks. The in-plane ordering in the liquid Al layers adjacent to both AlN{0 0 0 1} substrates is, however, weak.
3.3 Interfacial Interactions at L-Al/AlN{0 0 0 01}
Recent study revealed that substrate chemistry influences prenucleation at liquid-metal/solid-substrate interfaces.[8] Electronic structure study also showed that at liquid-metal/oxide interfaces, charge transfer occurs from the terminating metal atoms to the outmost oxygen atoms, which hinders the atomic ordering of the nearby liquid metal atoms.[10,11,37,39,41] Here, we apply the Bader’s charge analysis[50] to the electron density distributions at the interfaces from first-principles electronic structure calculations. The obtained charges at the atomic sites are plotted in Figure 7.

(Color-online)Charges at the atomic sites at the L-Al/AlN{0 0 0 1} interfaces. The dotted red lines represent the terminating Al layers
At the substrates, all the N atoms are negatively charged with a gain of 2.35 e/N and the Al lose 2.35 e/Al, corresponding to its high ionicity.[13] Meanwhile, the charge transfer value (2.35 e/atom) is smaller than the ionic model (3.0 e/atom), indicating ionic/covalent dual nature in AlN. The Al atoms away from interfaces are electronically neutral.
Interestingly, the Al atoms/ions adjacent to the two AlN{0 0 0 1} substrates exhibit different charging behaviors. The Al atoms near S2 are partially charged with an average of +0.52 e/Al for the terminating Al layer and about +0.10 e/Al for the 1st Al layer (right side in Figure 7). The sum of charges (about 0.63 e/Al) is close to the one quarter of charge at the Al in AlN (2.35 e/Al). This agrees with that the terminating Al atoms at S2 has one N neighbor (Figure 6(b)). The interfacial charge transfer limits to one or two liquid layers, indicating it local nature. This is due to the screening effect of the free electrons in liquid Al.
At the S1 interface, the terminating Al atoms are positively charged with an averaged valence of +1.96 e/Al, higher than three quarters of the charge in bulk (2.35*(3/4) = 1.76e). Moreover, the Al at the 1st and 2nd layers are negatively charged with a gain of 0.31 e/Al and 0.08 e/Al, respectively. This indicates that some of the electrons at the terminating Al layer transfer to the nearby Al atoms. The different charging at the terminating Al layers is one of the causes for the variation of atom numbers at the liquid Al layers at the L-Al/AlN{0 0 0 1} interfaces.
4 Discussion
Recent studies showed four factors influencing prenucleation at a liquid/solid interface: temperature (T), lattice misfit (f), chemistry of substrate (ΔH)) and atomic roughness of the substrate (R).[6,7,8,10] The chemical factor between liquid metal and substrates can be assessed by the charges at the atomic sites. The present AIMD simulations and electronic structure calculations for the L-Al/AlN{0 0 0 1} interfaces provide an opportunity to address this issue. Table I lists the lattice misfits, chemistry and geometry of the terminating metal layers, and the resultant prenucleation of the L-Al/AlN{0 0 0 1} interfaces with comparison with the related L-Al/alumina interfaces simulated at 1000 K.[10,11] The results for the ideal L-Al/s-Al interface[8] are included for comparison.
The lattice misfit between AlN{0 0 0 1} and α-Al{1 1 1} at its melting temperature is about −7 pct, which absolute value is higher than those at the L-Al/oxide interfaces (Table I). The terminating Al atoms/ions at L-Al/AlN{0 0 0 1} form flat peaks. They are positively charged with 1.96 e/Al at S1 and 0.52 e/Al at S2. The layering at the L-Al/AlN{0 0 0 1} interfaces is high (five Al layers), which is just lower than the idealized case (6 Al layers),[8] indicating lattice misfit and charging have a minor impact on layering. This agrees with the previous work for the pinned substrate.[7,8,51] On the other hand, the lattice misfits between Al and alumina substrates are positive and moderate (3.6 pct and 5.6 pct between Al/γ-Al2O3 and Al/α-Al2O3 respectively), while the terminating Al layers contain out-of-plane atomic displacements and atomic vacancies, being atomically rough.[10,11,39] Consequently, the layering and the atomic ordering at these interfaces is notably weaker, indicating that atomic roughness hinders both atomic layering and in-plane ordering at a liquid-metal/solid-substrate interface.[33]
As shown Table I, the in-plane coefficient of the terminating Al layers at the L-Al/AlN{0 0 0 1}S1 is high (0.41) whereas S(z) ~ 0.0 for the 1st Al layer. Meanwhile at the L-Al/AlN{0 0 0 1}S2, S(z) = 0.18 for the terminating Al layer and S(z) = 0.03 for the 1st Al layer. This indicates that the combination of pronounced lattice misfit and charging at the interfaces deteriorate the in-plane ordering at the interfaces. Table I shows that overall prenucleation at L-Al/AlN{0 0 0 1} is more pronounced than that at the L-Al/alumina interfaces. This indicates that the atomic roughness at terminating Al layer at the L-Al/Al2O3 interfaces overtakes the large lattice misfit at L-Al/AlN{0 0 0 1}.
The early stage solidification model showed that particles being less potent require larger driving forces (lower cooling temperature) to nucleate in nearby liquid.[5,6,16] The more pronounced prenucleation at the L-Al/AlN{0 0 0 1} interfaces than that at the L-Al/alumina interfaces indicates that the nucleation temperature of the liquid Al adjacent to the AlN particles is higher than those near the oxide particles.[5,6,16] Thus, under the same condition, nucleation at AlN occurs first. If the particle density of AlN in Al liquid is higher or close to that of alumina, they shall behave as potential nucleation sites. This might be realized by nitriding of Al-liquids during the liquid-dealing and casting. The experiments have found that the prepared AlN/Al composites containing well distributed AlN nano-partciles produced by means of nitriding of Al-liquids exhibit improved mechanical performances.[2,19,20,21] This topic deserves further investigation.
Here we discuss the competence between the native AlN and oxide particles as potential nucleation sites in liquid Al alloys. As mentioned before, the atomic roughness of the alumina oxide substrates means less potency to nucleate Al-based alloys. Consequently, the required nucleation temperature by aluminum oxide particles is low and might be even lower than the grain initiation temperature.[5,6,9] As soon as the grain initiation temperature is reached, heterogeneous nucleation and following grain initiation occurs almost simultaneously. Then one expects that a large fraction of particles may act as nucleation and grain initiation sites, and thus the grain-initiation occurs explosively. Strong grain inoculation occurs and fine particles in the cast alloys are expected if the density of the oxide particles is high enough with a uniform distribution.[5,6,9] In such cases, the more potent particles, such as AlN are harmful to the grain-refinement and their numbers should be minimized during casting.
The obtained information is helpful to understand the interaction between liquid Al to AlN particles, and further to enrich our understanding of heterogeneous nucleation,[5,6,16,52,53] as well as for AlN-containing composites[2,19,20,21,22] and ceramic-metal welding.[27]
5 Conclusions
We investigated prenucleation at the interfaces between liquid Al and the dipolar AlN{0 0 0 1} substrates using the ab initio molecular dynamics simulation and electronic band-structure calculations techniques. The study revealed formation of an ordered Al layer terminating the AlN substrates. Structurally, the terminating Al layers are coupled to the substrate bulk and can be considered as an extension of the AlN substrates. The liquid Al atoms adjacent to the substrates form flat layers. Chemically, the Al atoms terminating the substrates are positively charged. Moreover, charging of the 1st Al layer is unusual: The Al at 1st layer at S1 are negatively charged, whereas those at S2 are positively charged. There is strong layering but weak in-plane ordering due to the combination of the interfacial charging and the pronounced lattice misfit.
References
M. Yan, P. Yu, G.B. Schafffer, and M. Qian: Acta Mater., 2010, vol. 17, pp. 5667–74.
S. Fale, A. Likhite, and J. Bhatt: Trans. Ind. Inst. Met., 2013, vol. 66, pp. 265–71.
F. Wang and Z. Fan: Metall. Mater. Trans. A., 2019, vol. 50A, pp. 2519–26.
Z.P. Que and C.L. Mendis: Metall. Mater. Trans. A., 2021, vol. 52A, pp. 553–59.
Z. Fan, Heterogeneous nucleation, grain initiation and grain refinement of Mg-alloys, Proc. The 11th international conference on magnesium alloys and their applications, 24-27 July 2018, Beaumont Estate, Old Windsor, UK. Ed. By Z. Fan and C. Mendis, 2018, p 7.
Z. Fan, F. Gao, B. Jiang, and Z.P. Que: Sci. Rep., 2020, vol. 10, p. 9448.
H. Men and Z. Fan: Metall. Mater. Trans. A., 2018, vol. 49A, pp. 2766–77.
C.M. Fang, H. Men, and Z. Fan: Metall. Mater. Trans. A., 2018, vol. 49A, pp. 6231–42.
Z. Fan: Metal. Mater. Trans. A., 2013, vol. 44A, pp. 1409–18.
C.M. Fang and Z. Fan: Comput. Mater. Sci., 2020, vol. 171, p. 109258.
C.M. Fang, S. Yasmin, and Z. Fan: J. Phys. Comm., 2021, vol. 5, p. 015007.
W.M. Yim and R.J. Paff: J. Appl. Phys., 1974, vol. 45, pp. 1456–57.
C.M. Fang and R.A. de Groot: J. Phys. Condens. Matter., 2007, vol. 19, p. 386223.
P.W. Tasker: Philos. Mag. A., 1979, vol. 39, pp. 119–36.
M.S. Miao, A. Janotti, and C.G. Van de Walle: Phys. Rev. B., 2009, vol. 80, p. 155319.
K.F. Kelton and A.L. Greer: Nucleation in condensed matter: applications in materials and biology, Pergamon Materials Series, Elsevier, Oxford, 2010.
C. Toy and W.D. Scott: J. Mater. Sci., 1997, vol. 32, pp. 3243–48.
C. Borgonovo and M.M. Maklouf: Metall. Mater. Trans. A., 2016, vol. 47A, pp. 5125–35.
S. Fale, A. Likhite, and J. Bhatt: Trans. Ind. Inst. Met., 2014, vol. 67, pp. 841–49.
S. Fale, A. Likhite, and J. Bhatt: Trans. Ind. Inst. Met., 2015, vol. 68, pp. 291–97.
S.L. Pramod, S.R. Bakshi, and B.S. Murty: J. Mater. Eng. Perform., 2015, vol. 24, pp. 2185–07.
C. Borgonovo, D. Apelian, and M.M. Makhlouf: JOM., 2011, vol. 63, pp. 57–64.
C.M. Montesa, N. Shibata, T. Tohei, and Y. Ikuhara: J. Mater. Sci., 2011, vol. 46, pp. 4392–96.
J. Fritsch, O.F. Sankey, K.E. Schmidt, and J.B. Page: Phys. Rev. B., 1998, vol. 57, pp. 15360–71.
J.H. Cao, Y. Liu, and X.S. Ning: Materials., 2018, vol. 11, p. 775.
K. Choudhary, T. Liang, K. Mathew, B. Revard, A. Chernatynskiy, S.R. Phillpot, R.G. Henning, and S.B. Sinnott: Comput. Mater. Sci., 2016, vol. 113, pp. 80–87.
L. Mouradoff, T. Tristant, J. Desmaison, J.C. Labbe, M. Desmaison-Brut, and R. Rezakhanlou: Corr. Adv. Ceram., 1996, vol. 113, pp. 177–85.
S. Ogata and H. Kitagawa: Comput. Mater. Sci., 1999, vol. 15, pp. 435–40.
A. Kumamoto, N. Shibata, K.-I. Nayuki, T. Tohei, N. Terasaki, Y. Nagamoto, T. Nagase, K. Akiyama, Y. Kuromitsu, and Y. Ikuhara: Sci. Rep., 2016, vol. 6, p. 22936.
A. Hashibon, J. Adler, M.W. Finnis, and W.D. Kaplan: Comput. Mater. Sci., 2002, vol. 24, pp. 443–52.
H. Men and Z. Fan: Comput. Mater. Sci., 2014, vol. 85, pp. 1–7.
R. Yan, W.Z. Sun, S.D. Ma, R.L. Davidchak, N. Di Pasquale, Q.J. Zhai, T. Jin, and H.B. Dong: Comput. Mat. Sci., 2018, vol. 155, pp. 136–43.
B. Jiang, H. Men, and Z. Fan: Comput. Mater. Sci., 2018, vol. 153, pp. 73–81.
J.S. Wang, A.P. Horsfield, P.D. Lee, and P. Brommer: Phys. Rev. B., 2010, vol. 82, p. 14420.
J.S. Wang, A.P. Horsfield, U. Schwingenschlögl, and P.D. Lee: Phys. Rev. B., 2010, vol. 82, p. 144203.
D. Wearing, A.P. Horsfielf, W.W. Xu, and P.D. Lee: J. Alloys Compds., 2016, vol. 664, pp. 460–68.
S.H. Oh, Y. Kauffmann, C. Scheu, and W.D. Kaplan: Rühle, Ordered liquid aluminium at the sapphire. Sci., 2005, vol. 310, pp. 661–63.
R. Yan, W.Z. Sun, S.D. Ma, T. Jing, and H.B. Dong: Comput. Mater. Sci., 2020, vol. 174, p. 109489.
S.D. Ma, A.J. Brown, R. Yan, R.L. Davidchack, P.B. Howes, C. Nicklin, Q.J. Zhai, T. Jing, and H.B. Dong: Commun. Chem., 2019, vol. 2, p. 1.
S. Ma, Y. Rui, J. Tao, and H. Dong: Metals., 2018, vol. 8, p. 521.
C.M. Fang and Z. Fan: Metal. Mater. Trans. A., 2020, vol. 51A, pp. 6318–26.
J. Kang, J. Zhu, C. Curtis, D. Blake, G. Glatzmaier, Y.-H. Kim, and S.-H. Wei: Phys. Rev. Lett., 2012, vol. 108, p. 226105.
J.W. Arblaster: Selected values of the crystallographic properties of the elements, ASM International, Materials Park, 2018.
G. Kresse and J. Furthmüller: Comput. Mater. Sci., 1996, vol. 6, pp. 15–50.
P.E. Blöchl: Phys. Rev. B., 1994, vol. 50, pp. 17953–78.
J.P. Perdew, K. Burke, and M. Ernzerhof: Phys. Rev. Lett., 1996, vol. 77, pp. 3865–68.
H.J. Monkhorst and J.D. Pack: Phys. Rev. B., 1976, vol. 13, pp. 5188–92.
L.E. Hintzsche, C.M. Fang, T. Watts, M. Marsman, G. Jordan, G.M.W.P.E. Lamers, A.W. Weeber, and G. Kresse: Phys. Rev. B., 2012, vol. 86, p. 235204.
W. Kohn: Overview of density functional theory, in Density functional theory, NATO ASI series (Series B: Physics), vol. 337, E.K.U. Gross and R.M. Dreizler, eds., Springer, Boston, 1995.
R.F.W. Bader: Chem. Rev., 1991, vol. 91, pp. 893–928.
Z. Fan, H. Men, Y. Wang, and Z.P. Que: Metals., 2021, vol. 11, p. 478.
M.A. Easton, M. Qian, A. Prasad, and D.H. StJohn: Curr. Opin. Solid State Mater. Sci., 2016, vol. 20, pp. 13–24.
A.L. Greer: J. Chem. Phys., 2016, vol. 145, p. 21104.
Acknowledgments
We thank very much Prof. Brain Cantor for beneficial discussion. Financial support from EPSRC (UK) under Grant No. EP/N007638/1 is gratefully acknowledged.
Conflict of interest
The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, C.M., Fan, Z. Atomic Ordering at the Interfaces Between Liquid Aluminum and Polar AlN{0 0 0 1} Substrates. Metall Mater Trans A 53, 2040–2047 (2022). https://doi.org/10.1007/s11661-022-06646-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-022-06646-w