
Abstract
Diabetes mellitus is a disease of dysregulated blood glucose homeostasis. The current pandemic of diabetes is a significant driver of patient morbidity and mortality, as well as a major challenge to healthcare systems worldwide. The global increase in the incidence of diabetes has prompted researchers to focus on the different pathogenic processes responsible for type 1 and type 2 diabetes. Similarly, increased morbidity due to diabetic complications has accelerated research to uncover pathological changes causing these secondary complications. Albuminuria, or protein in the urine, is a well-recognised biomarker and risk factor for renal and cardiovascular disease. Albuminuria is a mediator of pathological abnormalities in diabetes-associated conditions such as nephropathy and atherosclerosis. Clinical screening and diagnosis of diabetic nephropathy is chiefly based on the presence of albuminuria. Given the ease in measuring albuminuria, the potential of using albuminuria as a biomarker of cardiovascular diseases is gaining widespread interest. To assess the benefits of albuminuria as a biomarker, it is important to understand the association between albuminuria and cardiovascular disease. This review examines our current understanding of the pathophysiological mechanisms involved in both forms of diabetes, with specific focus on the link between albuminuria and specific vascular complications of diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) has long been recognised as a serious challenge to health, due to its ability to affect almost every organ in the human body. Diabetes is broadly classified into two subtypes: type 1 (T1DM) and type 2 (T2DM), with differing clinical phenotypes that require distinct therapeutic management. Both forms of diabetes are associated with an increased risk of cardiovascular complications that can have a significant impact on patient morbidity and mortality. Albuminuria is one of the most commonly assessed clinical parameters in diabetic patients. A number of simple dipstick testing methods are available in current practice to measure protein levels in the urine. Albumin is a tightly regulated component in the circulation, with the kidney playing an important role in maintaining albumin homeostasis. The role of albuminuria as a potential driver as well as a biomarker of diabetic complications has been increasingly recognised. Given the potential of albuminuria as a marker of diabetic vascular complications, this review will first summarise T1DM and T2DM, followed by the pathogenic processes linking albuminuria and cardiovascular complications of diabetes.
Diabetes Mellitus
DM is a chronic endocrine condition that has reached pandemic levels worldwide. The disease is a growing public health concern, with 1 in 11 people worldwide affected by the condition [1]. Symptoms commonly associated with the onset of diabetes include increased urination (polyuria), increased thirst (polydipsia) and unintentional weight loss leading to increased appetite (polyphagia). Patients with diabetes have also reported slower wound healing, increased risk of acquiring infection, fatigue and blurred vision [2]. Clinically, DM is diagnosed in symptomatic patients when blood or urine sugar levels are elevated. A series of biochemical tests aid in confirming the diagnosis of diabetes in patients presenting with some of the symptoms listed above. In symptomatic patients, a fasting glucose concentration of ≥ 7 mmol/L (126 mg/dL) on two separate occasions is considered to be diagnostic for diabetes [3]. Haemoglobin (Hb) A1c has become widely used in clinical practice because it reflects long-term (3 months) glycaemic control [4,5,6]. A HbA1c reading of more than 48 mmol/mol (6.5%) supports the diagnosis of DM [3]. Based on clinical symptoms and age of onset, diabetes is broadly classified into T1DM and T2DM which account for approximately 10% and 90% of all diabetic cases respectively [2, 7]. Other forms of diabetes such as gestational diabetes and genetic forms of diabetes such as mature onset of diabetes in the young (MODY) also exist but will not be the focus of this review.
Type 1 Diabetes
T1DM is caused by an auto-immune-mediated destruction of insulin-producing β-cells within the pancreatic islets of Langerhans [2]. Early studies have described diverse aetiologies for T1DM including genetic predisposition, environmental influences and immunological responses [8,9,10,11,12]. Many T1DM patients develop auto-antibodies against insulin and other antigens that contribute to the immune-mediated destruction of pancreatic β-cells. A number of cellular death pathways such as apoptosis and necrosis of pancreatic β-cells have been described in T1DM pathophysiology [13, 14]. Rojas et al. (2018) identified intrinsic and extrinsic apoptotic pathways which are regulated by the B cell lymphoma (Bcl)-2 and tumour necrosis factor (TNF) family of ligands respectively [15]. Experiments conducted by Thomas et al. have, however, questioned the significance of the TNF ligand, FAS, in mediating β-cell apoptosis as very few FAS producing cells were detected in T1DM mice models [16]. In contrast to apoptosis, necrotic and necroptotic processes initiate a cascade of immunological reactions in T1DM islets. Necroptosis or programmed necrosis is also facilitated by TNF signalling, which results in the synthesis of reactive oxygen species (ROS) [15]. In addition to this, calcium has been identified as a potent signalling modulator in necroptosis [17]. Genome-wide analysis has established interferon and tyrosine kinase 2 genes play a key role in eliciting apoptotic and necrotic pathways pancreatic β-cells [18].
Type 2 Diabetes
The International Diabetes Federation estimates that there are currently 463 million people between the ages of 20 and 79 years living with diabetes [19]. Half of these patients (232 million) remain undiagnosed, and the number of diabetes patients is predicted to increase to 700 million by 2045 [19]. There is a strong genetic predisposition in both T1DM and T2DM pathogenesis. Mutations in HLA and HNF1B genes are strongly associated with the pathogenesis of T1DM and T2DM respectively [20, 21]. Currently, obesity is the strongest environmental risk factor associated with T2DM [22,23,24]. In the majority of individuals, obesity leads to insulin resistance and causes compensatory hyperplastic transformation of β-cells leading to hyperinsulinaemia that strives to overcome insulin resistance in peripheral tissues such as liver, muscle and fat. However, in T2DM patients, these early hypertrophic changes in pancreatic islets are overtaken by atrophic changes and a gradual loss of β-cells and insulin production. It is thought that obesity results in the de-differentiation of pancreatic α-cells into β-cells. Increased metabolic dysregulation induced by diabetes suppresses the transcription factor FOXO1. As a consequence, pancreatic β-cells undergo trans-differentiation into other types of pancreatic endocrine cells which leads to reduced pancreatic β-cell mass [24, 25].
Reduced insulin action in the liver, muscle and fat are key drivers of insulin resistance leading to hyperglycaemia in T2DM [26, 27]. Hyperglycaemia-induced metabolic dysregulation causes glucotoxicity which triggers pathogenic changes in T2DM [28]. Glucotoxicity increases reactive oxygen species (ROS) production resulting in glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibition which subsequently decreases the antioxidant capabilities of β-cells [29]. Lack of glucose utilisation by adipose tissue promotes free fatty acid production which has been shown to induce programmed cell death of β-cells [30, 31]. Recently, the role of islet amyloid protein in β-cell death has been described [32, 33]. This polypeptide is released along with insulin in response to increased blood sugar levels. In diabetic conditions, there is increased secretion of the amyloid protein as a result of enhanced insulin secretion. Amyloid then accumulates within the endoplasmic reticulum of β-cells dysregulating cell cycle mediating transcription factors. As a result, apoptosis of β-cells is initiated within the pancreas, thereby reducing pancreatic β-cell mass, leading to insulin deficiency and hyperglycaemia [27,28,29].
Diabetic Vascular Complications
The pathogenic changes described above can lead to dysregulated glucose homeostasis which can damage endothelial and other cell functions within the vasculature. These unregulated glucose levels are a major cause of chronic diabetic complications, which significantly increases mortality associated with DM [34,35,36]. Based on the organ and size of blood vessels affected, complications of DM are classified as macrovascular or microvascular. The heart, brain and peripheral vasculature are grouped as macrovascular complications of diabetes, which are major causes of death in people living with diabetes. For example, women living with diabetes have a 5-fold higher risk of myocardial infarction, an 8-fold higher risk of stroke and a 40-fold higher risk of requiring a foot amputation due to peripheral arterial disease (PAD) [37]. Cardiovascular conditions such as myocardial infarction (MI), heart failure and cardiomyopathies have been reported to be the major contributors of mortality in people living with diabetes [38]. Atherosclerosis is an inflammatory vascular condition which in the diabetic milieu accelerates cardiovascular and neurovascular diseases associated with macrovascular diabetic complications [39, 40].
The eye and the kidney are organs affected by diabetic microvascular complications. Diabetic eye disease or retinopathy is the leading cause of blindness worldwide [41]. Diabetic kidney disease, or nephropathy (DN), is the leading cause of end-stage renal disease (ESRD) in the working-age population [42]. Up to 50% of patients are diagnosed with diabetes with at least one diabetic complication evident (usually some degree of diabetic retinopathy). Both diabetic retinopathy and nephropathy are classified according to a series of stages, indicating the relative severity of the disease in each patient (Tables 1 and 2). These microvascular complications significantly impact the quality of life (e.g. visual loss) and reduce survival (e.g. ESRD). Between 25 and 40% of individuals with diabetes develop DN. Up to one-third of people with DN will progress to ESRD and diabetic kidney disease is now the leading cause of ESRD globally [42]. DN is diagnosed clinically when protein is detected in the urine and/or there is evidence of chronic deterioration in kidney function [3]. Given the poor prognosis for patients receiving chronic dialysis, together with the shortage of available donor kidneys for transplanation, a better understanding of the pathogenesis of DN and the role that albumin plays in this process is needed.
Albuminuria
The concept of measuring albumin in urine was first suggested by a German scientist, Hermann Senator, in the nineteenth century [43]. Albuminuria has been identified as the most sensitive marker for abnormal kidney function [44]. Stage I DN is characterised by microalbuminuria levels of 30–300-mg/24 h urine (Table 3). Measuring albumin levels from a 24-h urine sample has been considered the gold standard [2]. In 2012, The Kidney Disease: Improving Global Outcomes (KDIGO) organisation classified albuminuria as normal (< 30 mg/24 h), moderately increased/microalbuminuria (30–300 mg/24 h) and severely increased/macroalbuminuria (> 300 mg/24 h) (Table 3; [43]). A urine dipstick is the easiest and quickest way of detecting macroalbuminuria but it is a rather insensitive test for quantifying urine protein concentration. The urinary albumin-to-creatinine ratio (UACR) is convenient and equivalent to the gold standard technique for calculating ACR, with ratios < 3, 3–30 and > 30 mg/mmol being classified as normal, moderate and severe albuminuria respectively (Table 3; [2]). In addition to being a predictive marker of DN, albuminuria has also been found to be a useful prognostic marker for cardiovascular disease (CVD) in diabetic patients [45, 46]. To understand the intricate relationship between albuminuria and diabetic cardiovascular complications, it is first important to understand the function of albumin in the physiological setting.
Albumin Homeostasis
Albumin is a small negatively charged protein synthesised mainly by the liver, contributing to 10% of overall protein production in the body. Albumin makes up ~ 75% of normal plasma colloid oncotic pressure and 50% of plasma protein content [47]. Albumin plays a key role in transporting molecules such as fatty acids, metals, bilirubin and hormones such as glucocorticoids in the plasma to their target cells [47]. Many commonly used drugs such as benzodiazepines, warfarin and cloxacillin bind to a number of different sites on albumin in plasma [48]. Albumin production is a highly regulated process modulated by various physiological and pathological conditions [49, 50]. Under normal conditions, levels of urinary albumin should be very low, with < 30 mg detectable in a 24-h urine sample (Table 3). A number of hormones, including glucocorticoids and insulin, are key regulators of albumin homeostasis [51]. In diabetes, the levels of these hormones are dysregulated, thereby leading to abnormal albumin metabolism. Breakdown of albumin occurs in most organs of the body, but mainly the muscle and skin [52]. The fractional catabolic rate of albumin is another important regulatory factor of albumin concentration in pathophysiologic states such as diabetes. In healthy individuals, renal tubular epithelial cells re-absorb the majority of albumin that is filtered by glomeruli and tubular cells can also degrade albumin via lysosomal proteolysis [53, 54]. In contrast, other authors report that albumin is protected from lysosomal degradation in tubular epithelial cells and is recycled intact to the circulation via tubular transcytosis [55].
Albumin and the Kidney
Under normal conditions, the glomerulus, an intricate vascular structure in the kidney, limits the transport of albumin from blood to urine [54]. The glomerular filtration barrier is made up of glomerular endothelial cells on the blood vessel side, the glomerular basement membrane and podocytes with interdigitating foot processes on the urine side of the barrier [56]. Studies by Ryan and Karnovsky have identified the endothelial layer of the glomerular filtration apparatus to be the most effective barrier to albumin filtration [57, 58]. Multiple hypotheses have been postulated how this is achieved [57,58,59,60,61]. One of the first theories explored the size of the endothelial filtration pore as a preventative system for albumin filtration [62]. Gagliardini et al. carried out imaging of glomerular epithelial filtration slit using scanning electron microscopy in physiological and proteinuric conditions [63]. In this study, larger glomerular barrier pores were observed in mice with progressive renal fibrosis and diabetic nephropathy, thereby suggesting a positive correlation between albuminuria and dysregulation of glomerular pore size. Furthermore, Comper et al. examined the potential role of electrical charge as a barrier to albumin filtration [64]. Albumin and the glomerular vascular membrane are both negatively charged, suggesting that electrostatic repulsion of albumin molecules away from the vessel wall is an important component of barrier function [52]. This hypothesis was supported by a number of other reports suggesting that electrostatic repulsion was a major factor limiting the filtration of albumin from blood to urine [65,66,67]. In diseases such as diabetic nephropathy, the integrity of the glomerular filtration barrier is compromised by chronic hyperglycaemia, leading to increased albumin filtration into the urine (and detectable albuminuria).
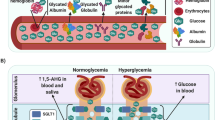
The other nephron structure involved in regulating albumin excretion is the renal tubule (Fig. 1). In normal health, a small amount of albumin is filtered by glomeruli, and the majority of the filtered albumin is subsequently reabsorbed by the proximal and distal tubules [56]. A number of cellular transport processes, such as endocytosis and transcytosis, enable tubular epithelial cells to retrieve albumin from the glomerular filtrate [68,69,70]. In the proximal convoluted tubule, albumin binds to the megalin-cubilin receptor, resulting in endocytosis of the protein into vesicles which is either stored or degraded within the cytoplasm [69]. Many animal studies [69,70,71,72,73] have been carried out with results supporting this theory. An experimental study measured the levels of megalin and cubilin between diabetic and non-diabetic mice and identified the downregulation of these receptor proteins in diabetic mice [74]. Another study showed similar results and went on to demonstrate increased endocytosis and expression of renal megalin with insulin treatment in the diabetic group compared with the control group [75].

Schematic diagram showing how megalin and cubilin may contribute to albumin filtration from the blood vessels into the urine via proximal tubule epithelial cells. The key for each of the indicated molecules is shown at the base of the figure
Albuminuria and Diabetic Vascular Complications
Albuminuria and Diabetic Nephropathy
Glomerulosclerosis and tubulointerstitial fibrosis are two major pathological hallmarks of DN [76]. Glomerulosclerosis is thought to occur as the initial insult in DN, followed by a progressive, irreversible fibrosis or scarring of the kidney tubules that occurs in parallel with the slow decline in renal function [76]. A variety of pathological mechanisms which contribute to diabetic kidney damage have been identified [42]. Injury to the glomerular filtration barrier leads to compromised filtration by the glomerulus which has been linked to the onset of renal fibrosis, potentially due to increased levels of albumin in the urinary filtrate [77]. Hyperglycaemia-induced haemodynamic dysregulation promotes increased production of endogenous vasodilators such as nitric oxide, insulin-like growth factor (IGF) and vascular endothelial growth factor (VEGF) [78]. Hyperglycaemia also promotes cellular oxidative stress resulting in the formation of advanced glycation end products (AGEs) [79]. These metabolic changes induce pathological changes in the glomerulus such as mesangial expansion, formation of Kimmelstiel-Wilson nodules and glomerular basement membrane (GBM) thickening [44, 76]. Damage to the glomerular basement membrane increases albumin excretion by increasing the pore size and disturbing the electrical homeostasis of the GBM.
Chronic hyperglycaemia is also associated with irreversible changes to renal tubular structure and function [75, 80, 81]. Transforming growth factor-beta (TGFβ) is an important cytokine driving the pathogenic changes in both diabetic glomerulus and kidney tubules [82]. TGFβ causes the expansion of glomerular mesangial cells, thickening of the GBM and loss of filtration barrier integrity [83]. TGFβ is also implicated in tubulointerstitial fibrosis, where activated myofibroblasts generated from vascular pericytes, tubular epithelial cells and resident fibroblasts secrete extracellular matrix proteins such as collagen IV and fibronectin that contribute to scar formation [84,85,86].
Recent experiments performed by Mori et al. analysed albumin excretion in diabetic megalin gene knockout mice [87]. A significant increase in albumin excretion was detected in diabetic megalin knockout mice compared with diabetic wild type [87], suggesting that lower megalin expression reduced the capacity of the kidney tubular epithelial cells to sequester albumin in the kidney filtrate. The potential role of megalin in potentiating TGFβ-induced epithelial-mesenchymal transition (EMT) has also been studied. TGFβ was used to stimulate EMT in epithelial renal cells and reduced levels of megalin were detected in these cells and these changes were reversed when the cells were treated with a TGFβ inhibitor [88, 89]. A meta-analysis of data from genome-wide association studies (GWAS) has identified a mutation in the cubilin-encoding gene CUBN that is associated with microalbuminuria in European and African populations [90].
Additionally, a post hoc analysis of data from the Diabetes Control and Complications Trial (DCCT) and Epidemiology of Diabetes Interventions and Complications (EDIC) study also confirmed an association between CUBN mutations and an increased risk of developing microalbuminuria [91, 92]. Increased cubilin expression was also observed in T1DM mice with cubilin mRNA levels negatively correlated with albuminuria [93]. These and other reports highlight the significance of the megalin-cubilin receptor pathway in tubular cell albumin uptake and the pathogenesis of diabetic kidney disease [72, 73, 78, 87, 94].
Albuminuria and Atherosclerosis
The causative link between albuminuria and CVD has been extensively studied, yet the pathophysiological mechanism has not been clearly identified [95,96,97,98]. The most commonly accepted theory is the STENO hypothesis, which states that glomerular dysfunction is reflective of extensive vascular damage throughout the body [99]. In diabetes, processes such as endothelial dysfunction and chronic inflammation lead to the development of angiopathies in many organs. Microalbuminuria has been associated with an increased risk of developing subclinical atherosclerosis in the Mexican population, with another study reporting the same in elderly patients living with diabetes [100, 101]. The intimal-media thickness (IMT) is an early indicator of developing carotid atherosclerosis [102]. Zhang et al. assessed IMT using high-frequency ultrasonography and reported a strong correlation between microalbuminuria and IMT [103]. Multiple studies have examined the link between inflammatory cytokines and atherosclerosis accelerated by diabetes [104,105,106]. Cytokines such as interleukin-6 (IL-6) and tumour necrosis factor alpha (TNFα), along with acute-phase proteins like C-reactive protein (CRP), were shown to promote inflammatory processes within the vasculature leading to endothelial damage and dysfunction [107,108,109,110]. Despite strong evidence supporting the link between microalbuminuria and diabetes-induced atherosclerosis, recent evidence has identified microalbuminuria to be an independent risk factor for atherosclerosis even in the absence of diabetes [111, 112]. A cross-sectional study performed by Kimura et al. studied 1756 non-diabetic men with normal estimated glomerular filtration rate (eGFR), and the results indicated that patients with high-normal albuminuria had a significantly higher risk of IMT and developing atherosclerotic plaques [113]. A recent gene analysis study has identified mutations in the APOL1 gene to be associated with this outcome, but further studies are needed to validate this finding [114].
Albuminuria and Myocardial Infarction
Myocardial infarction (MI) is a cardiovascular condition which is commonly associated with microalbuminuria. Multiple clinical trials targeting different population groups have concluded that increased albumin excretion is associated with an increased risk of MI [106, 115,116,117,118]. Secondary analysis of data from the TRACER study has reported microalbuminuria was associated with an increased risk of MI and cardiovascular mortality [118]. Another study of Mexican patients with diabetes also identified a higher number of major adverse cardiovascular events associated with increased albuminuria levels [117]. These findings were further supported by the REGARDS study which examined persons with diabetes with and without coronary heart disease (CHD). The results concluded that higher urinary albumin-to-creatinine ratio was associated with greater risk of incident of heart disease in black versus white patients > 45 years in the USA [119]. These conclusions suggest that albuminuria is a strong biomarker for CHD development and mortality. The underlying pathogenic mechanisms are similar to those described for atherosclerosis, with aberrant endothelial signalling and low-grade vascular inflammation within coronary arteries being key drivers of MI in people with diabetes. Therefore, control of albuminuria clearly has the potential to improve the outcomes of patients at risk from MI and other cardiovascular diseases.
Albuminuria and Heart Failure
Clinical studies have identified increased urinary albumin excretion among heart failure patients [118, 120,121,122,123]. A prospective study carried out in a cohort of patients with well-characterised heart failure and preserved ejection fraction showed increased urinary albumin excretion was associated with enhanced remodelling of the right and left ventricles [124]. During follow-up of this cohort, raised urinary albumin excretion was also associated with systolic dysfunction (assessed by echocardiography) [125]. Post hoc analysis of the SAVOR-TIMI 53 trial cohort also identified increased hospitalisation for heart failure among patients with microalbuminuria [126]. Another study recruited 100 asymptomatic people living with diabetes and compared their cardiac extracellular volume fraction with urinary microalbumin levels. A strong positive association was observed between these two measurements, suggesting diffuse cardiac fibrosis is the underlying pathogenic mechanism involved in heart failure development among patients with persistent microalbuminuria. The glomerulus is one of the first vascular structures to be affected by diabetes resulting in microalbuminuria. As stated previously, glomerular damage is considered to be indicative of a more widespread vascular injury including the heart.
Though a direct correlation between albuminuria and cardiac fibrosis has not been established, a number of clinical trials have identified existing diabetic medications which may reduce albuminuria and potentially attenuate fibrosis [127,128,129,130,131]. Sodium-glucose transport protein 2 (SGLT2) inhibitors are widely used medications for diabetes management which also has been shown to reverse microalbuminuria. Early studies concluded that this class of drugs are beneficial in the treatment of heart failure [132]. Dapagliflozin is a SGLT2 inhibitor drug with cardioprotective properties [129, 133]. Data from the DApagliflozin on renal outcomes and cardiovascular mortality in PAtients with Chronic Kidney Disease (DAPA-CKD) study suggested that dapagliflozin may be an efficient therapy for heart failure, though more complete analysis of data from this trial is required before any substantive conclusions can be made [129]. Additionally, the potential role of renin-angiotensin-aldosterone system blockade in halting heart failure development among diabetic patients has also been highlighted [120]. Angiotensin-converting enzyme inhibitors (ACE-I) are currently the first-line treatment for heart failure [134]. The BENEDICT (BErgamo NEphrologic DIabetes Complications Trial) and ADVANCE (Action in Diabetes and Vascular Disease-preterAx and diamicroN Controlled Evaluation) trial concluded that ACE-I help in delaying microalbuminuria onset in persons with T2DM, leading researchers to postulate that this may be an additional mechanism underlying the effectiveness of ACE-I in heart failure treatment, in addition to its vasodilatory action [135, 136].
Albuminuria and Peripheral Arterial Disease
Peripheral arterial disease (PAD) is caused by large and small blood vessel damage resulting in ischaemia and necrosis of the extremities (typically the legs and feet). Diabetes and atherosclerosis are major causes of PAD, and the loss of circulation can lead to critical limb ischaemia and may require lower limb amputation. The potential link between albuminuria and PAD in patients living with diabetes has been investigated, with different studies providing conflicting evidence. A cross-sectional study performed among 1197 patients with T2DM showed that PAD was associated with albuminuria [137]. This was supported by the Multi-Ethnic Study of Atherosclerosis (MESA) study which showed that the risk of developing PAD was almost doubled in patients presenting with albuminuria [138]. Despite the positive correlation between albuminuria and PAD among diabetic subjects, the results from the National Health and Nutrition Examination Survey 1999–2004 showed that this association was also observed among non-diabetic subjects. Non-invasive methods to identify PAD, such as the use of Doppler ankle-brachial pressure index, are more practical for general population studies of the prevalence of PAD than gold standard angiography [139]. A number of pathological mechanisms have been suggested to contribute to the development of PAD among patients with albuminuria. In the diabetic milieu, endothelial dysfunction produces an excess of reactive oxygen species (ROS) which subsequently leads to an increase in platelet activity and vascular smooth muscle proliferation. This phenomenon is believed to play a significant role in the development and progression of PAD [140].
Conclusion
Diabetes-associated complications are major contributors to patient morbidity and mortality, as well as to the global healthcare burden. Current and emerging treatments for diabetic vascular complications target a range of processes aimed at preserving vascular function and maintaining tissue homeostasis. Albuminuria has been identified to be a useful diagnostic marker of DN, indicative of damage to the glomerular filtration barrier. The hyperglycaemic state observed in diabetes results in aberrant cellular signalling causing damage to the kidneys, eyes, heart and other organs. The kidneys can be severely affected in diabetes, with vascular and tubular structures undergoing detrimental pathological changes as a result of hyperglycaemia which in due course leads to albuminuria. The damage to the glomerular capillaries can be considered to be representative of the widespread vascular damage evoked in the diabetic milieu.
The increased CVD risk conferred by albuminuria has been well established in the literature. The pathogenic changes induced by albuminuria can accelerate the development of many cardiovascular conditions such as atherosclerosis, MI and heart failure. A summary of potential mechanisms of albuminuria as a result of micro- and macrovascular diabetic complications is shown in Fig. 2. Despite substantial evidence, the significance of albuminuria in diabetic complications has yet to be included in many clinical trials. The complex pathological processes involved have given researchers a wealth of biochemical pathways to study further. Continued and future support for pre-clinical and clinical studies into the significance of albuminuria will be fundamental in successfully translating animal studies into improved vascular outcomes for patients living with diabetes.

Summary of mechanisms leading to diabetic kidney disease and albuminuria
References
Organization WH. Diabetes [Internet]. [cited 2020 Jun 1]. https://www.who.int/news-room/fact-sheets/detail/diabetes
Diagnosis and Classification of Diabetes Mellitus. Diabetes Care [Internet]. 2014;37:S81 LP-S90. http://care.diabetesjournals.org/content/37/Supplement_1/S81.abstract
NICE guideline. Type 2 diabetes in adults : management. NICE Guide 28. NICE Guid [Internet]. 2019;NG28:1–57. https://www.nice.org.uk/guidance/ng28/resources/type-2-diabetes-in-adults-management-1837338615493
Mostafa SA, Coleman RL, Agbaje OF, Gray AM, Holman RR, Bethel MA. Modelling incremental benefits on complications rates when targeting lower HbA1c levels in people with type 2 diabetes and cardiovascular disease. Diabet Med . John Wiley & Sons, Ltd (10.1111). 2018;35:72–7.
Kweka B, Lyimo E, Kidola J, Filteau S, Friis H, Manjurano A, et al. Validity of HbA1c in diagnosing diabetes among people with sickle cell trait in Tanzania. Blood. 2019;134:4852.
Mottalib A, Salsberg V, Mohd-Yusof B-N, Mohamed W, Carolan P, Pober DM, et al. Effects of nutrition therapy on HbA1c and cardiovascular disease risk factors in overweight and obese patients with type 2 diabetes. Nutr J. 2018;17:42.
O’Neill S. Us, diabetes and a lot of facts and stats. Diabetes UK. 2019:1–48 www.diabetes.org.uk.
Rojas J, Bermudez V, Palmar J, Martínez MS, Olivar LC, Nava M, et al. Pancreatic beta cell death: novel potential mechanisms in diabetes therapy. J Diabetes Res. 2018;2018:1–19.
Usmani-Brown S, Perdigoto AL, Lavoie N, Clark P, Korah M, Rui J, et al. Β cell responses to inflammation. Mol Metab. 2019;27:S104–13.
Paschou SA, Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. On type 1 diabetes mellitus pathogenesis. Endocr Connect. 2017/11/30. Bioscientifica Ltd; 2018;7:R38–R46. https://www.ncbi.nlm.nih.gov/pubmed/29191919.
van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev United States. 2011;91:79–118.
La Torre D, Lernmark A. Immunology of beta-cell destruction. Adv Exp Med Biol United States. 2010;654:537–83.
Wilcox NS, Rui J, Hebrok M, Herold KC. Life and death of β cells in type 1 diabetes: a comprehensive review. J Autoimmun. 2016/03/24. 2016;71:51–8. https://www.ncbi.nlm.nih.gov/pubmed/27017348.
Tomita T. Apoptosis of pancreatic β-cells in Type 1 diabetes. Bosn J basic Med Sci [Internet]. Association of Basic Medical Sciences of Federation of Bosnia and Herzegovina; 2017;17:183–193. https://www.ncbi.nlm.nih.gov/pubmed/28368239.
Rojas J, Bermudez V, Palmar J, Martinez MS, Olivar LC, Nava M, et al. Pancreatic beta cell death: novel potential mechanisms in diabetes therapy. J Diabetes Res. England; 2018;2018:9601801.
Thomas HE, Darwiche R, Corbett JA, Kay TW. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. J Immunol United States. 1999;163:1562–9.
Lee M-S, Chang I, Kim S. Death effectors of beta-cell apoptosis in type 1 diabetes. Mol Genet Metab United States. 2004;83:82–92.
Marroqui L, Dos Santos RS, Floyel T, Grieco FA, Santin I, Op de Beeck A, et al. TYK2, a candidate gene for type 1 diabetes, modulates apoptosis and the innate immune response in human pancreatic beta-cells. Diabetes. United States; 2015;64:3808–3817.
Internation Diabetes Federation. IDF diabetes Atlas Ninth. Dunia IDF. 2019.
De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C, Foti DP. Type 2 diabetes mellitus and cardiovascular disease: genetic and epigenetic links [Internet]. Front. Endocrinol. . 2018. p. 2. https://www.frontiersin.org/article/10.3389/fendo.2018.00002
Zeng C-P, Lin X, Peng C, Zhou L, You H-M, Shen J, et al. Identification of novel genetic variants for type 2 diabetes, childhood obesity, and their pleiotropic loci. J Hum Genet. 2019/02/28. 2019;64:369–77. https://www.ncbi.nlm.nih.gov/pubmed/30816286.
Perng W, Oken E, Dabelea D. Developmental overnutrition and obesity and type 2 diabetes in offspring. Diabetologia. 2019;62:1779–88.
Grarup N, Moltke I, Andersen MK, Dalby M, Vitting-Seerup K, Kern T, et al. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat Genet. 2018;50:172–4.
Koffert J, Ståhle M, Karlsson H, Iozzo P, Salminen P, Roivainen A, et al. Morbid obesity and type 2 diabetes alter intestinal fatty acid uptake and blood flow. Diabetes, Obes Metab. John Wiley & Sons, Ltd (10.1111); 2018;20:1384–90.
Yagihashi S, Inaba W, Mizukami H. Dynamic pathology of islet endocrine cells in type 2 diabetes: β-Cell growth, death, regeneration and their clinical implications. J Diabetes Investig. 2015/10/15. John Wiley and Sons Inc.; 2016;7:155–65. https://www.ncbi.nlm.nih.gov/pubmed/27042265.
DeFronzo RA. In: Bonora E, DeFronzo RA, editors. Pathogenesis of type 2 diabetes mellitus BT - diabetes epidemiology, genetics, pathogenesis, diagnosis, prevention, and treatment. Cham: Springer International Publishing; 2018. p. 181–253.
Merino J, Leong A, Liu C-T, Porneala B, Walford GA, von Grotthuss M, et al. Metabolomics insights into early type 2 diabetes pathogenesis and detection in individuals with normal fasting glucose. Diabetologia. 2018;61:1315–24.
Diiorio P, Jurczyk A, Yang C, Racki WJ, Brehm MA, Atkinson MA, et al. Hyperglycemia-induced proliferation of adult human beta cells engrafted into spontaneously diabetic immunodeficient NOD-Rag1null IL2rgammanull Ins2Akita mice. Pancreas. United States; 2011. p. 1147–1149.
Leibowitz G, Kaiser N, Cerasi E. β-Cell failure in type 2 diabetes. J Diabetes Investig. Blackwell Publishing Ltd; 2011;2:82–91. https://www.ncbi.nlm.nih.gov/pubmed/24843466.
Palomer X, Pizarro-Delgado J, Barroso E, Vázquez-Carrera M. Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab. 2018;29:178–90 http://www.sciencedirect.com/science/article/pii/S1043276017301704.
Mardinoglu A, Gogg S, Lotta LA, Stančáková A, Nerstedt A, Boren J, et al. Elevated plasma levels of 3-hydroxyisobutyric acid are associated with incident type 2 diabetes. EBioMedicine. 2018;27:151–5 http://www.sciencedirect.com/science/article/pii/S2352396417304899.
Bellotti V, Corazza A, Foglia B, Novo E, Simons JP, Mangione PP, et al. Amyloid damage to islet β-cells in type 2 diabetes: hypoxia or pseudo-hypoxia? bioRxiv. 2019;810747. http://biorxiv.org/content/early/2019/10/24/810747.abstract
Hsu Y-H, Chen Y-W, Wu M-H, Tu L-H. Protein glycation by glyoxal promotes amyloid formation by islet amyloid polypeptide. Biophys J. 2019;116:2304–13 http://www.sciencedirect.com/science/article/pii/S0006349519304151.
Papatheodorou K, Banach M, Bekiari E, Rizzo M, Edmonds M. Editorial. Complications of diabetes 2017. 2018;2018.
Forbes JM, Cooper ME. Mechanisms of diabetic complications 2019;137–188.
Volpe CMO, Villar-Delfino PH, dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9:119.
Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther United States. 2008;88:1254–64.
Xia F, Liu G, Shi Y, Zhang Y. Impact of microalbuminuria on incident coronary heart disease, cardiovascular and all-cause mortality: a meta-analysis of prospective studies. Int J Clin Exp Med. e-Century Publishing Corporation; 2015;8:1–9. https://www.ncbi.nlm.nih.gov/pubmed/25784968.
E KJ, Farah K, Shelley B, Karishma R, Jaume A, Xiaochao W, et al. Abstract 414: Local artery wall inflammation overrides systemic inflammation in diabetes-accelerated atherosclerosis. Arterioscler Thromb Vasc Biol. American Heart Association; 2018;38:A414–A414.
Tatjana J, Tessa B, Emily B, Alexandra Q, Yoscar O, Felix Z, et al. Abstract 13598: Neutrophil extracellular traps (NETs) promote macrophage inflammation and contribute to diabetes-impaired atherosclerosis regression. Circulation. American Heart Association; 2019;140:A13598–A13598.
Wang W, Lo ACY. Diabetic retinopathy: pathophysiology and treatments. Int J Mol Sci. 2018;19.
Magee C, Grieve DJ, Watson CJ, Brazil DP. Diabetic nephropathy: a tangled web to unweave. Cardiovasc Drugs Ther. United States; 2017;31:579–592.
Gansevoort RT, Ritz E. Hermann senator and albuminuria—forgotten pioneering work in the 19th century. Nephrol Dial Transplant. 2008;24:1057–62.
Warren AM, Knudsen ST, Cooper ME. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opin Ther Targets England. 2019;23:579–91.
Herzog CA, Asinger RW, Berger AK, Charytan DM, Díez J, Hart RG, et al. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2011;80:572–86. http://www.sciencedirect.com/science/article/pii/S0085253815551053
Feakins BG, McFadden EC, Farmer AJ, Stevens RJ. Standard and competing risk analysis of the effect of albuminuria on cardiovascular and cancer mortality in patients with type 2 diabetes mellitus. Diagnostic Progn Res. 2018;2:13.
Peralta R. What is the function of albumin in the human body? Medscape.
Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol. 2005;353:38–52 http://www.sciencedirect.com/science/article/pii/S0022283605008855.
Rothschild MA, Oratz M, Schreiber SS. Albumin metabolism. Gastroenterology. Elsevier; 1973;64:324–337.
Miyamoto H, Tao X, Kohzuma T, Ohnishi A. Influences of anemia, kidney disease, thyroid dysfunction, and liver disease on the ratio of glycated albumin to hemoglobin A1c. J Diabetes Sci Technol. 2018/04/05. SAGE Publications; 2018;12:1082–1083. https://www.ncbi.nlm.nih.gov/pubmed/29619893.
Tommerdahl KL, Brinton JT, Timb V, Nadeau KJ, Zeitler P, Chan CL. Screening for cystic fibrosis–related diabetes and prediabetes—evaluating 1,5-anhydroglucitol, fructosamine, glycated albumin, and hemoglobin A1c. Diabetes. 2018;67 1531-P. http://diabetes.diabetesjournals.org/content/67/Supplement_1/1531-P.abstract.
Levitt DG, Levitt MD. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med. Dove Medical Press; 2016;9:229–255. https://www.ncbi.nlm.nih.gov/pubmed/27486341.
Tojo A. The role of the kidney in protein metabolism : the capacity of tubular lysosomal proteolysis in nephrotic syndrome. Kidney Int Elsevier Masson SAS. 2013;84:861–3.
Moeller MJ, Tenten V. Renal albumin filtration: alternative models to the standard physical barriers. Nat Rev Nephrol. 2013;9:266–77.
Tenten V, Menzel S, Kunter U, Sicking E, Roeyen CRC Van, Sanden SK, et al. Albumin is recycled from the primary urine by tubular transcytosis 2013;1966–1980.
Birn H, Christensen EI. Renal albumin absorption in physiology and pathology. Kidney Int. Elsevier Masson SAS; 2006;69:440–449.
Ryan GB, Karnovsky MJ. Distribution of endogenous albumin in the rat glomerulus: role of hemodynamic factors in glomerular barrier function. Kidney Int United States. 1976;9:36–45.
Ryan GB. The glomerular sieve and the mechanisms of proteinuria. Aust N Z J Med [Internet]. John Wiley & Sons, Ltd (10.1111); 1981;11:197–206.
Satchell S. The role of the glomerular endothelium in albumin handling. Nat Rev Nephrol. 2013;9:717–25.
Mima A, Yasuzawa T, King GL, Ueshima S. Obesity-associated glomerular inflammation increases albuminuria without renal histological changes. FEBS Open Bio. John Wiley & Sons, Ltd; 2018;8:664–70.
Rippe B. What is the role of albumin in proteinuric glomerulopathies? Nephrol Dial Transplant [Internet]. 2004;19:1–5.
Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol med (Berl). Germany; 1998;76:172–183.
Gagliardini E, Conti S, Benigni A, Remuzzi G, Remuzzi A. Imaging of the porous ultrastructure of the glomerular epithelial filtration slit. J Am Soc Nephrol. 2010/10/28. American Society of Nephrology; 2010;21:2081–9. https://pubmed.ncbi.nlm.nih.gov/21030599
Comper WD. Is there equivalency of intact albuminuria and albumin peptideuria in nephrotic states? Kidney Int. Elsevier; 2013;84:1050.
Schlöndorff D, Wyatt CM, Campbell KN. Revisiting the determinants of the glomerular filtration barrier: what goes round must come round. Kidney Int. Elsevier; 2017;92:533–536.
Suh JH, Miner JH. The glomerular basement membrane as a barrier to albumin. Nat Rev Nephrol. 2013;9:470–7.
Aoki S, Saito-Hakoda A, Yoshikawa T, Shimizu K, Kisu K, Suzuki S, et al. The reduction of heparan sulphate in the glomerular basement membrane does not augment urinary albumin excretion. Nephrol Dial Transplant. 2017;33:26–33.
Dickson LE, Wagner MC, Sandoval RM, Molitoris BA. The proximal tubule and albuminuria: really! J Am Soc Nephrol. 2014/01/09. American Society of Nephrology; 2014;25:443–53. https://www.ncbi.nlm.nih.gov/pubmed/24408874
Kryvenko V, Vadász I. The role of CD36 in endothelial albumin transcytosis. 2019;738–9.
Lin NYC, Homan KA, Robinson SS, Kolesky DB, Duarte N, Moisan A, Lewis JA Renal reabsorption in 3D vascularized proximal tubule models. Proc Natl Acad Sci U S A. 2019/03/04. National Academy of Sciences; 2019;116:5399–5404. https://www.ncbi.nlm.nih.gov/pubmed/30833403.
Amsellem S, Gburek J, Hamard G, Nielsen R, Willnow TE, Devuyst O, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21:1859 LP – 1867. http://jasn.asnjournals.org/content/21/11/1859.abstract
Gekle M, Knaus P, Nielsen R, Mildenberger S, Freudinger R, Wohlfarth V, et al. Transforming growth factor-β1 reduces megalin- and cubilin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. J Physiol. John Wiley & Sons, Ltd (10.1111); 2003;552:471–81.
Thrailkill KM, Nimmo T, Bunn RC, Cockrell GE, Moreau CS, Mackintosh S, et al. Microalbuminuria in type 1 diabetes is associated with enhanced excretion of the endocytic multiligand receptors megalin and cubilin. Diabetes Care. 2009;32:1266 LP – 1268. http://care.diabetesjournals.org/content/32/7/1266.abstract
Figueira MF, Castiglione RC, de Lemos Barbosa CM, Ornellas FM, da Silva Feltran G, Morales MM, et al. Diabetic rats present higher urinary loss of proteins and lower renal expression of megalin, cubilin, ClC-5, and CFTR. Physiol Rep. John Wiley & Sons, Ltd; 2017;5:e13335.
Bryniarski MA, Yee BM, Jaffri I, Chaves LD, Yu JA, Guan X, Ghavam N, Yacoub R, Morris ME Increased megalin expression in early type 2 diabetes: role of insulin-signaling pathways. Am J Physiol Physiol [Internet]. American Physiological Society; 2018;315:F1191–F1207.
Schena FP, Gesualdo L. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. United States; 2005;16 Suppl 1:S30–3.
Podgorski P, Konieczny A, Lis L, Witkiewicz W, Hruby Z. Glomerular podocytes in diabetic renal disease. Adv Clin Exp Med Poland. 2019;28:1711–5.
Lin Y-C, Chang Y-H, Yang S-Y, Wu K-D, Chu T-S. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. 2018;117:662–75 http://www.sciencedirect.com/science/article/pii/S0929664617308033.
Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia Germany. 2001;44:1957–72.
Comper WD. Kidney Int. Elsevier; 2018;93:1014. Megalin/cubilin has a minor role in the proximal tubular cell uptake of filtered albumin.
Giraud-billoud M, Fader CM, Agüero R, Ezquer F, Ezquer M. Diabetic nephropathy , autophagy and proximal tubule protein endocytic transport : a potentially harmful relationship. 2018;
Kato M, Yuan H, Xu Z-G, Lanting L, Li S-L, Wang M, et al. Role of the Akt/FoxO3a pathway in TGF-β1–mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J Am Soc Nephrol. 2006;17:3325 LP – 3335. http://jasn.asnjournals.org/content/17/12/3325.abstract
Wang S, Zhou Y, Zhang Y, He X, Zhao X, Zhao H, et al. Roscovitine attenuates renal interstitial fibrosis in diabetic mice through the TGF-β1/p38 MAPK pathway. Biomed Pharmacother. 2019;115:108895. http://www.sciencedirect.com/science/article/pii/S0753332219303701
Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. United States. 2015;21:989–97.
Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med United States. 2015;21:998–1009.
Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre J V, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. United States; 2010;176:85–97.
Mori KP, Yokoi H, Kasahara M, Imamaki H, Ishii A, Kuwabara T, et al. Increase of total nephron albumin filtration and reabsorption in diabetic nephropathy. J Am Soc Nephrol. United States. 2017;28:278–89.
Xiao Y, Liu J, Peng Y, Xiong X, Huang L, Yang H, et al. GSTA3 attenuates renal interstitial fibrosis by inhibiting TGF-beta-induced tubular epithelial-mesenchymal transition and fibronectin expression. PLoS One. 2016;11:1–15.
Tian M, Tang L, Wu Y, Beddhu S, Huang Y. Adiponectin attenuates kidney injury and fibrosis in deoxycorticosterone acetate-salt and angiotensin II-induced CKD mice. Am J Physiol Ren Physiol. 2018;315:F558–71.
Boger CA, Chen M-H, Tin A, Olden M, Kottgen A, de Boer IH, et al. CUBN is a gene locus for albuminuria. J Am Soc Nephrol United States. 2011;22:555–70.
Predictors of the development of microalbuminuria in patients with type 1 diabetes mellitus: a seven-year prospective study. The Microalbuminuria Collaborative Study Group. Diabet Med. England; 1999;16:918–925.
Vergouwe Y, Soedamah-Muthu SS, Zgibor J, Chaturvedi N, Forsblom C, Snell-Bergeon JK, et al. Progression to microalbuminuria in type 1 diabetes: development and validation of a prediction rule. Diabetologia. 2009/11/04. Springer-Verlag; 2010;53:254–262. https://pubmed.ncbi.nlm.nih.gov/19908023
Feng M, Zhang Z, Fu P, Huang S, Yang Y, Su K, et al. Expression level of cubilin in the rat model of diabetic nephropathy. Sichuan Da Xue Xue Bao Yi Xue ban. China; 2006;37:738–741.
Li W, Yang S, Qiao R, Zhang J. Potential value of urinary exosome-derived let-7c-5p in the diagnosis and progression of type ii diabetic nephropathy. Clin Lab. 2018;64:709–18. http://europepmc.org/abstract/MED/29739042
Oellgaard J, Gæde P, Persson F, Rossing P, Parving H, Pedersen O. Journal of Diabetes and Its Complications Application of urinary proteomics as possible risk predictor of renal and cardiovascular complications in patients with type 2-diabetes and microalbuminuria. J Diabetes Complications. Elsevier Inc.; 2018;32:1133–40.
Karalliedde J, Viberti G. Hypertension and microalbuminuria: risk factors for cardiovascular disease in diabetes. Curr Hypertens Rep. 2005;7:1–2.
Weir MR. Microalbuminuria and Cardiovascular Disease. 2007;581–90.
Nijpels G, Stehouwer CDA, Højlund K, Nolan JJ, Balkau B, Dekker JM. Insulin sensitivity and albuminuria : the RISC study 2014;37:1597–1603.
Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis Diabetologia Germany. 1989;32:219–26.
Medina-Urrutia A, Juárez-Rojas JG, Posadas-Sánchez R, Jorge-Galarza E, Cardoso-Saldaña G, Vargas-Alarcón G, et al. Microalbuminuria and its association with subclinical atherosclerosis in the Mexican mestizo population: the GEA study. Rev Investig Clin organo del Hosp Enfermedades la Nutr. Mexico; 2016;68:262–268.
Cao JJ, Barzilay JI, Peterson D, Manolio TA, Psaty BM, Kuller L, et al. The association of microalbuminuria with clinical cardiovascular disease and subclinical atherosclerosis in the elderly: the cardiovascular health study. Atherosclerosis. 2006;187:372–7 http://www.sciencedirect.com/science/article/pii/S0021915005006052.
Bernard S, Sérusclat A, Targe F, Charrière S, Roth O, Beaune J, et al. Incremental predictive value of carotid ultrasonography in the assessment of coronary risk in a cohort of asymptomatic type 2 diabetic subjects. diabetes Care. 2005;28:1158 LP – 1162. http://care.diabetesjournals.org/content/28/5/1158.abstract
Zhang Y-H, Gao Y, Mao X, Shang J, Su B-L. Assessment of carotid atherosclerosis in type 2 diabetes mellitus patients with microalbuminuria by high-frequency ultrasonography. Migdalis I, editor. Int J Endocrinol. Hindawi Publishing Corporation; 2013;2013:819584.
Vadivelu R, Vijayvergiya R. Panvascular risk factor – diabetes. Cor Vasa [Internet]. 2018;60:e18–29 http://www.sciencedirect.com/science/article/pii/S0010865017301704.
Bornfeldt KE, Kramer F, Batorsky A, Choi J, Hudkins KL, Tontonoz P, et al. A novel type 2 diabetes mouse model of combined diabetic kidney disease and atherosclerosis. Am J Pathol. 2018;188:343–52. http://www.sciencedirect.com/science/article/pii/S0002944017304765
Duivenvoorden R. Atherosclerosis and diabetic nephropathy BT - diabetic nephropathy: pathophysiology and clinical aspects. In: Roelofs JJ, editor. Vogt L, editors. Cham: Springer International Publishing; 2019. p. 357–72.
Palmas W, Ma S, Jacobs DRJ, Arnett D, Jackson S, Olson J, et al. Ethnicity and sex modify the association of serum c-reactive protein with microalbuminuria. Ethn Dis United States. 2008;18:324–9.
Kitatani N, Taniguchi A, Fukushima M, Nakai Y, Nagasaka S, Ohgushi M, et al. Association of serum resistin with TNF system activity in Japanese type 2 diabetic patients. Diabetes Metab. France; 2007. p. 156–157.
Norbert H, Ruth S-U, Ralf D, Joon-Keun P, Fatimunnisa Q, Maren W, et al. Vascular endothelial cell–specific NF-κB suppression attenuates hypertension-induced renal damage. Circ Res. American Heart Association; 2007;101:268–276.
Nguyen TT, Alibrahim E, Islam FMA, Klein R, Klein BEK, Cotch MF, et al. Inflammatory, hemostatic, and other novel biomarkers for diabetic retinopathy: the multi-ethnic study of atherosclerosis. Diabetes Care United States. 2009;32:1704–9.
Junichi I, E. GM, P. NR, C. CM, R. LL, Shinichi U, et al. Hemoglobin, albuminuria, and kidney function in cardiovascular risk: the ARIC (Atherosclerosis Risk in Communities) Study. J Am Heart Assoc. American Heart Association; 2019;7:e007209.
Anuwatmatee S, Allison MA, Shlipak MG, McClelland RL, Kramer H, Tang S, et al. Relationship of fibroblast growth factor 21 with kidney function and albuminuria: multi-ethnic study of atherosclerosis. Nephrol Dial Transplant. 2018;34:1009–16.
Kimura T, Ueno T, Doi S, Nakashima A, Doi T, Ashitani A, Kawano R, Yamane K, Masaki T High-normal albuminuria is associated with subclinical atherosclerosis in male population with estimated glomerular filtration rate ≥60 mL/min/1.73 m2: A cross-sectional study. PLoS One. Public Libr Sci; 2019;14:e0218290. .
Mukamal KJ, Tremaglio J, Friedman DJ, Ix JH, Kuller LH, Tracy RP, et al. APOL1 genotype, kidney and cardiovascular disease, and death in older adults. Arterioscler Thromb Vasc Biol United States. 2016;36:398–403.
Mondesir FL, Brown TM, Muntner P, Durant RW. Diabetes , diabetes severity , and coronary heart disease risk equivalence : REasons for Geographic and Racial Differences in Stroke ( REGARDS ). Am Heart J. Elsevier Inc.; 2011;181:43–51.
Saleh M, Ambrose JA. Understanding myocardial infarction. F1000Research [Internet]. F1000 Research Limited; 2018;7:F1000 Faculty Rev-1378. https://www.ncbi.nlm.nih.gov/pubmed/30228871.
Tern MIS, Illalpando CLGO. Microalbuminuria as a predictor of myocardial infarction in a Mexican population : The Mexico City Diabetes Study 2005;68:34–39.
Åkerblom A, Clare RM, Lokhnygina Y, Wallentin L. Albuminuria and cardiovascular events in patients with acute coronary syndromes : results from the TRACER trial. Am Heart J. Elsevier Inc.; 178:1–8.
Gutiérrez OM, Khodneva YA, Muntner P, Rizk DV, McClellan WM, Cushman M, et al. Association between urinary albumin excretion and coronary heart disease in black vs white adults. JAMA. 2013;310:706–14.
Swoboda PP, Mcdiarmid AK, Erhayiem B, Ripley DP, Dobson LE, Garg P, et al. Diabetes mellitus, microalbuminuria, and subclinical cardiac disease: identi fi cation and monitoring of individuals at risk of heart failure 2017;1–11.
Page SEE. Albuminuria is independently associated with cardiac remodeling, abnormal right and left ventricular function, and worse outcomes in heart failure with preserved ejection fraction. JACC Hear Fail. Elsevier Inc. 2014;2:586–96.
Ohkuma T, Jun M, Chalmers J, Cooper ME, Hamet P, Harrap S, et al. Combination of changes in estimated GFR and albuminuria and the risk of major clinical outcomes. Clin J Am Soc Nephrol. 2019;14:862 LP – 872. http://cjasn.asnjournals.org/content/14/6/862.abstract
Boer IH De, Gao X, Cleary PA, Bebu I, Lachin JM, Molitch ME, et al. Article Albuminuria changes and cardiovascular and renal outcomes in type 1 diabetes : the DCCT / EDIC study. 2016;1969–77.
Senthil S, Brian C, J. SS, Inder A, L. RJ, Eileen O, et al. Prognostic value of albuminuria and influence of spironolactone in heart failure with preserved ejection fraction. Circ Hear Fail. American Heart Association; 2018;11:e005288.
Nayor M, Larson MG, Wang N, Santhanakrishnan R, Lee DS, Tsao CW, et al. The association of chronic kidney disease and microalbuminuria with heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail. John Wiley & Sons, Ltd; 2017;19:615–23.
Scirica BM, Mosenzon O, Bhatt DL, Udell AJ, Steg PG, McGuire DK, et al. Cardiovascular outcomes according to urinary albumin and kidney disease in patients with type 2 diabetes at high cardiovascular risk: observations from the SAVOR-TIMI 53 trial. JAMA Cardiol. 2018;3:155–63.
Lytvyn Y, Godoy LC, Scholtes RA, van Raalte DH, Cherney DZ. Mineralocorticoid antagonism and diabetic kidney disease. Curr Diab Rep. 2019;19:4.
P. LCS, Chanchal C, Vineeta A, Subodh V. SGLT-2 Inhibitors in heart failure: current management, unmet needs, and therapeutic prospects. j am heart assoc. American Heart Association; 2019;8:e013389.
Heerspink HJL, Stefansson B V, Chertow GM, Correa-Rotter R, Greene T, Hou F-F, et al. Rationale and protocol of the dapagliflozin and prevention of adverse outcomes in chronic kidney disease (DAPA-CKD) randomized controlled trial. Nephrol Dial Transplant. Oxford University Press; 2020;35:274–282. https://pubmed.ncbi.nlm.nih.gov/32030417
Groß M-L, Heiß N, Weckbach M, Hansen A, El-Shakmak A, Szabo A, et al. ACE-inhibition is superior to endothelin A receptor blockade in preventing abnormal capillary supply and fibrosis of the heart in experimental diabetes. Diabetologia. 2004;47:316–24.
Oellgaard J, Gæde P, Rossing P, Rørth R, Køber L, Parving H-H, et al. Reduced risk of heart failure with intensified multifactorial intervention in individuals with type 2 diabetes and microalbuminuria: 21 years of follow-up in the randomised Steno-2 study. Diabetologia. 2018;61:1724–33.
Karin R, Gemma F, Vlado P, D. SS, W. MK, dick de Z, et al. Canagliflozin and heart failure in type 2 diabetes mellitus. Circulation. American Heart Association; 2018;138:458–468.
Yoshihara F, Imazu M, Hamasaki T, Anzai T, Yasuda S, Ito S, et al. An exploratory study of dapagliflozin for the attenuation of albuminuria in patients with heart failure and type 2 diabetes mellitus (DAPPER). Cardiovasc Drugs Ther. 2018;32:183–90.
National Institute for Health and Care Excellence. Chronic heart failure in adults: diagnosis and management [Internet]. https://www.nice.org.uk/guidance/ng106
Heller SR. A summary of the ADVANCE trial. Diabetes Care. 2009;32:S357 LP-S361. http://care.diabetesjournals.org/content/32/suppl_2/S357.abstract
Remuzzi G, Macia M, Ruggenenti P. Prevention and treatment of diabetic renal disease in type 2 diabetes: the BENEDICT study. J Am Soc Nephrol. United States. 2006;17:S90–7.
Ito H, Takeuchi Y, Ishida H, Antoku S, Abe M, Mifune M, et al. High frequencies of diabetic micro- and macroangiopathies in patients with type 2 diabetes mellitus with decreased estimated glomerular filtration rate and normoalbuminuria. Nephrol Dial Transplant. 2009;25:1161–7.
Wattanakit K, Folsom AR, Criqui MH, Kramer HJ, Cushman M, Shea S, et al. Albuminuria and peripheral arterial disease: results from the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2008;201:212–6 http://www.sciencedirect.com/science/article/pii/S0021915008000191.
Wu C-K, Yang C-Y, Tsai C-T, Chiu F-C, Huang Y-T, Lee J-K, et al. Association of low glomerular filtration rate and albuminuria with peripheral arterial disease: the National Health and Nutrition Examination Survey, 1999–2004. Atherosclerosis. 2010;209:230–4 http://www.sciencedirect.com/science/article/pii/S0021915009007011.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature England. 2001;414:813–20.
Funding
PR is supported by the Pathological Society of Great Britain and Ireland. DPB is supported by MRC, NC3Rs, Invest NI, DfE of Northern Ireland and China Queen’s College Scholarship.
Author information
Authors and Affiliations
Contributions
PR wrote the article and designed the figures. DPB and APM provided editorial corrections and advice about structure and flow.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raja, P., Maxwell, A.P. & Brazil, D.P. The Potential of Albuminuria as a Biomarker of Diabetic Complications. Cardiovasc Drugs Ther 35, 455–466 (2021). https://doi.org/10.1007/s10557-020-07035-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-020-07035-4